
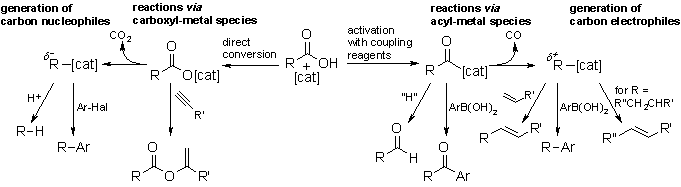
Carboxylic acids as synthetic equivalents to alkyl, aryl or acyl halides
One primary focus of our group is the development of cross-coupling reactions where the substrates are carboxylic acids instead of the ecologically questionable organyl halides usually employed. The carboxylic acids are activated in situ with coupling reagents, to allow an oxidative addition to transition metal catalysts. On the other hand, their metal salts can be converted into carbon nucleophiles via metal mediated decarboxylation.
Reviews:
| 1. | |
| 2. | |
| 3. | |
| 4. |
Decarboxylative cross-coupling reactions
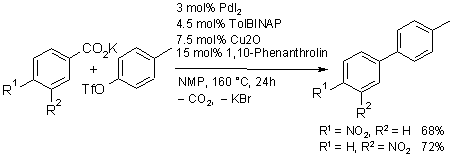
We developed a safe and convenient cross-coupling strategy for the synthesis of biaryls, commercially important structures often found in biologically active molecules. In contrast to traditional cross-couplings, which require the prior preparation of
organometallic reagents, we use a copper catalyst to generate the carbon nucleophiles in situ, via decarboxylation of easily
accessible arylcarboxylic acid salts. The scheme below depicts, how this new transformation turns simple carboxylic acidc salts into synthetic equivalents of valuable organometallic reagents, saving several steps in a synthetic sequence.
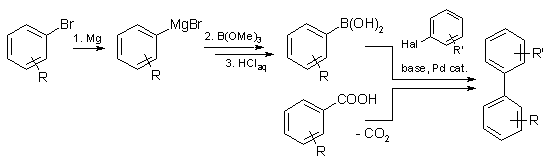
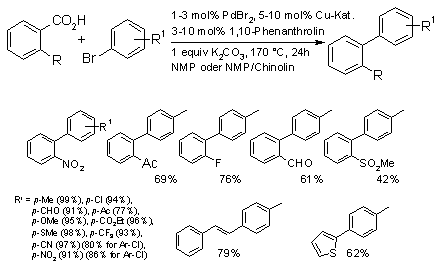
The decarboxylative biaryl synthesis is broadly applicable with respect to the aryl halide component and has successfully been
applied to a growing number of aromatic carboxylic acids, including ortho-substituted benzoic acids, heterocyclic carboxylates and cinnamic acid. Already at its early state of development, it opened up new opportunities for the industrial synthesis of high-value pharmaceutical intermediates.
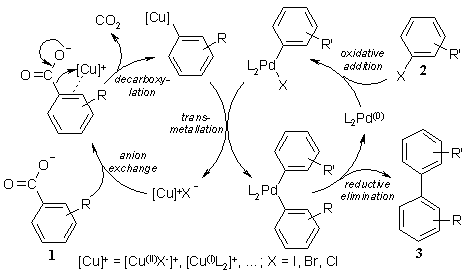
In our proposed mechanism, a decarboxylation catalyst, e.g. a copper or silver complex, initially coordinates to the carboxylate
oxygen, then shifts to the aryl p-system and inserts into the C–C(O) bond under extrusion of CO2 to form a stable aryl-copper
intermediate. A palladium catalyst then cross-couples this species with an aryl electrophile to form the desired biaryl and the
corresponding metal halide.
Whereas the first generation catalyst allowed the coupling of aryl iodides, bromides, and some electron-poor chlorides, the latest systems can smoothly convert even notoriously unreactive electron-rich aryl chlorides such as 4-chloroanisole. The main factors in this increased activity were optimized steric and electronic properties of the phosphine ligands.
When using aryl halides as coupling partners, the coupling of meta- and para-substituted benzoic acids has not yet been achieved in satisfactory yields, as the halide salts formed as byproducts in the cross coupling step compete with these carboxylates for coordination sites at the copper and thus interfere with the decarboxylation step. However, this structural limitation can be overcome when employing aryl triflates instead of halides, demonstrating that decarboxylative couplings are not intrinsically limited to a small range of carboxylates.
Another exciting development was the extension of the concept of decarboxylative cross-coupling to other substrate classes, as exemplified by a new ketone synthesis that draws on easily available potassium a-oxocarboxylates as sources of acyl nucleophiles in a coupling reaction with aryl bromides. The new reaction is broadly applicable to the synthesis of various aryl and heteroaryl ketones and compares favorably with traditional ketone syntheses via organometallic reagents.
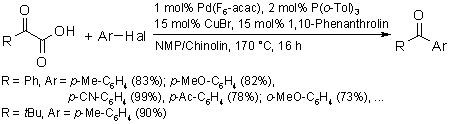
For mechanistic investigations of decarboxylative cross-coupling reactions see here.
Key references:
| 1. | |
| 2. | |
| 3. | |
| 4. | |
| 5. | |
| 6. | |
| 7. | |
| 8. | |
| 9. | |
| 10. | |
| 11. |
Ketone synthesis from carboxylic and boronic acids
The acylation of carbon nucleophiles with activated carboxlicy acid derivatives is an important C-C bond-forming reaction, which is extensively used in the synthesis of natural products and pharmaceutical compounds. However, carboxylic acids themselves can only be transformed into the corresponding ketones by reaction with highly reactive organometal compounds under rather elaborate conditions intolerant of most functional groups.
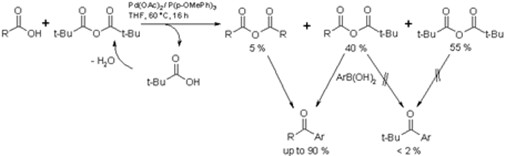
We have developed the first palladium(0)-catalyzed cross-coupling reaction between arylboronic acids and alkyl, vinyl or aryl carboxylic anhydrides producing the corresponding ketones. Furthermore, we elaborated a one-pot procedure, which allows the in situ generation of mixed anhydrides from the carboxylic acids and pivalic anhydride and their direct conversion to the ketones along with pivalic acid. Various arylketones were thus synthesized from the corresponding carboxylic and boronic acids in wet THF at 60 °C using a catalyst generated in situ from palladium acetate and tri-p-methoxyphenylphosphine. Even sensitive functionalities such as OH-groups are tolerated. Instead of pivalic anhydride, dimethyldicarbonate can also be used as activating agent.
Extensions of this methodology
We also developed an alternative procedure best used for small-scale applications e. g. in combinatorial chemistry. In this variant, the carboxylic acids are activated in situ by the commercially available coupling reagent DSC instead of pivalic anhydride.
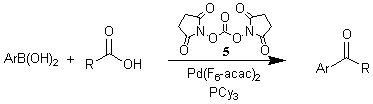
Key references:
| 1. | |
| 2. | |
| 3. | |
| 4. |
Salt-free Heck olefinations
Acyl-palladium complexes decarbonylate at elevated temperatures. This behavior was exploited in a new decarbonylative Heck olefination of carboxylic acids. In this process, aromatic carboxylic acids can be converted to vinyl arenes in the presence of di-tert-butyl dicarbonate.
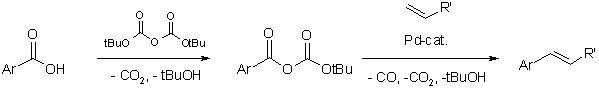
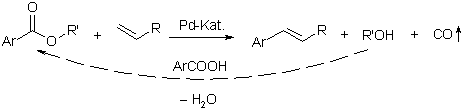
With especially developed palladium catalysts, even poorly reactive carboxylates were utilized as substrates for decarbonylative Heck olefinations. Hence, various esters of electron-deficient phenols were coupled with olefins to give the vinyl arenes, along with CO and the corresponding phenols. These were then recycled into the starting material in an esterification step with fresh carboxylic acid. Thus, it was experimentally demonstrated for the first time that the production of waste salts is avoidable in Heck olefinations.
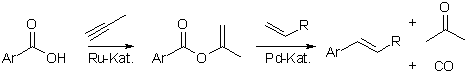
Further development of the catalysts enabled the decarbonylative Heck olefination of aryl isopropenoates to give the vinyl arenes, CO, and acetone. Combined with the formation of the isopropenyl esters from the carboxylic acids and propyne-a side product in oil refining-this also becomes a salt-free overall process. Besides CO, acetone is the only byproduct, and can be incinerated without any negative impact on the environment. Thus, a recycling of the byproduct is no longer necessary.
Key references:
| 1. | |
| 2. | |
| 3. |
Reduction of carboxylic acids to aldehydes
This approach led to the discovery of a selective palladium-catalyzed transfer hydrogenation of carboxylic acids to aldehydes. The acids are converted in situ into anhydrides using pivalic anhydride, and consequently reduced with sodium hypophosphite in the presence of a palladium catalyst.
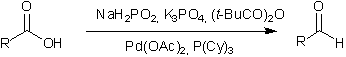
Key references:
| 1. | |
| 2. |
Decarbonylative eliminations
Pd-acyl-complexes of aliphatic carboxylic acids that bear b-hydrogens were found to undergo decarbonylation followed by facile b-hydride elimination at elevated conditions. We have exploited this behaviour in the development of a catalytic decarbonylative elimination reaction that allows the conversion of aliphatic carboxylic acids to alkenes under mild conditions.
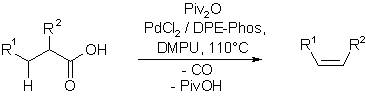
Key reference:
| 1. |
Decarbonylative Suzuki biaryl synthesis
In the presence of rhodium catalysts, even cross-coupling reactions can be performed under extrusion of carbon monoxide. This discovery set the stage for the development of a novel decarbonylative Suzuki coupling, that allows the synthesis of biaryls from aromatic carboxylic anhydrides and arylboroxines.
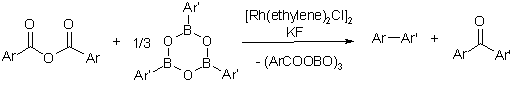
Key reference:
| 1. |