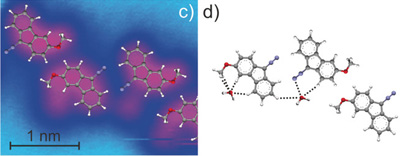
Welcome to Physical organic chemistry
News Highlights



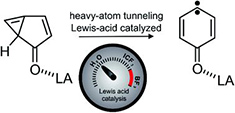
Heavy-atom quantum tunnelling catalysed with Lewis acids
Read the News Article in Chemistry World
Read our publication in Chemical Science



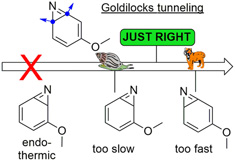
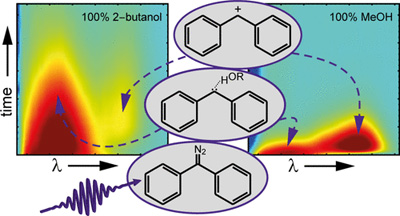

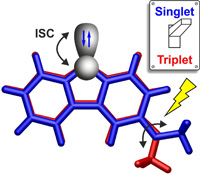



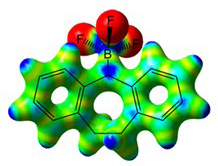
Our recent paper was mentioned in the magazine "Chemistry Views"
Read the article in Chemistry Views