Teilprojekt D: Prof. Dr. G. Jansen
Universität Duisburg-Essen
Quantum Chemical Calculations of Molecular Aggregates
It is the delicate interplay between electrostatic, induction, dispersion, and exchange interactions which results in the existence of small and large sized molecular aggregates such as gas phase clusters or molecular crystals. With the DFT-SAPT combination of symmetry-adapted perturbation theory for the treatment of intermolecular interactions and density functional theory for the calculation of the required molecular properties we dispose of an efficient tool for the precise characterization of the distance and orientation dependence of the various intermolecular forces.
This method was succesfully used to study the interactions between molecules possessing CH groups, π systems and lone electron pairs (acetylene, benzene, heteroaromates) and their consequences for the formation and the structure of molecular aggregates.
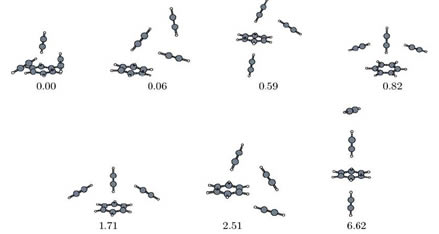
Various structures of the aggregate of one benzene and three acetylene molecules as obtained from model potentials determined with DFT-SAPT. Relative energies in kJ/mol.
The effects of the replacing CH with CF groups form a new focus of our investigations, as also the case in corresponding experimental work of other projects of FOR618 . DFT-SAPT and further quantum chemical methods are used to calculate interaction energies, to characterize the interactions in terms of individual energy contributions, and to derive analytical representations of potential energy surfaces for the interacting molecules. These then serve as the basis for molecular dynamics investigations of clusters and periodic systems and for the calculations of vibration-rotation-tunneling spectra of dimeric systems, thus enabling direct comparison with experiment.