Teilprojekt B: Prof. Dr. N. Doltsinis
Ruhr-Universität Bochum
Ab-initio Molekulardynamik-Simulationen wasserstoffverbrückter Systeme: von der Aggregationskinetik bis zur thermischen Umwandlung
Aggregate, Flüssigkeiten und Co-Kristalle bestehend aus schwach wechselwirkenden Molekülen sollen mithilfe von Ab Initio Molekulardynamik (AIMD) basierend auf der Dichtefunktionaltheorie (DFT) untersucht werden. DFT-Dispersionskorrekturen sollen implementiert und getestet werden, um eine verbesserte Beschreibung besonders schwacher molekularer Wechselwirkungen (z.B. in Co-Kristallen) zu erreichen und somit das Anwendungsfeld der AIMD bedeutend zu erweitern.
Um Hyperflächen Freier Energie im Rahmen der AIMD effizienter abtasten zu können sollen neue Methoden implementiert und existierende Methoden verbessert werden.
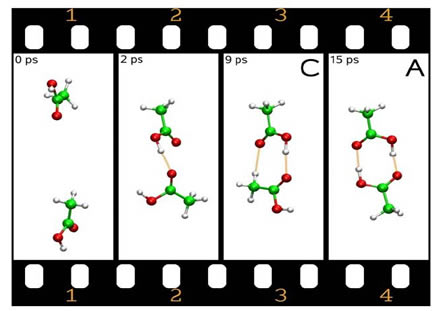
Schnappschüsse aus einer CP-MD Simulation der Dimerisierung von Essigsäure. Das Bild zeigt eine vorübergehende lokale Minimumsstruktur C, die zwei H-Brücken aufweist, eine CH...O und eine OH...O Brücke.
Ein besonderer Schwerpunkt der geplanten Anwendungen ist die (kinetisch kontrollierte) Aggregation kleiner Moleküle unter Matrixisolationsbedingungen und in ultrakalten Heliumtröpfchen unter Berücksichtigung der Molekül-Edelgas Wechselwirkungen. Dabei soll über die Dimerisierung hinaus auch die Bildung grösserer Komplexe untersucht werden. Auswirkungen der Substitution einzelner Atome (z.B. Deuterierung) auf die Produktverteilung der Aggregation sollen studiert werden. Die für die thermische Umlagerung der Aggregate entscheidenden freien Energielandschaften sollen in Abhängigkeit der Temperatur charakterisiert werden.