Fakultät für
Chemie
und Biochemie
Biochemie II
Biomolekulare NMR-Spektroskopie
Prof. Dr. Raphael Stoll
Gebäude NC 5/171
Universitätsstraße 150
D-44780 Bochum
Tel.: +49 234 32-25466
Fax: +49 234 32-05466
E-Mail: bionmr@rub.de







Medicinal Chemistry
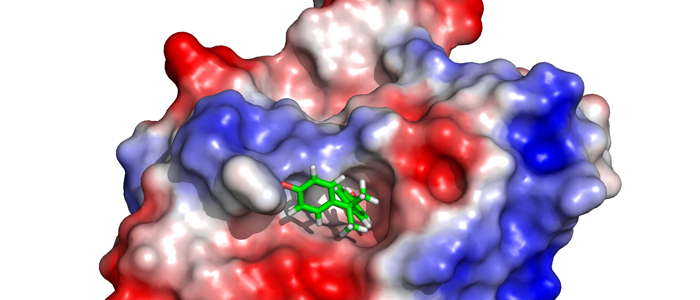
 The protein family of small GTPases controls cellular processes by acting as a binary switch between an active and an inactive state. The most prominent family members are H-Ras, N-Ras, and K-Ras isoforms, which are highly related and frequently mutated in cancer. Bisphenols are widespread in modern life because of their industrial application as plasticisers. Bisphenol A (BPA) is the best-known member and has gained significant scientific as well as public attention as an endocrine disrupting chemical, a fact that eventually led to its replacement. However, compounds used to replace BPA still contain the molecular scaffold of bisphenols. Here we show that BPA, BPAF, BPB, BPE, BPF, and an amine-substituted BPAF-derivate all interact with all GDP-bound Ras-Isoforms through binding to a common site on these proteins. NMR-, SOScat-, and GDI- assay-based data revealed a new bisphenol-induced, allosterically activated GDP-bound Ras conformation that define these plasticisers as Ras allosteric agonists.
The protein family of small GTPases controls cellular processes by acting as a binary switch between an active and an inactive state. The most prominent family members are H-Ras, N-Ras, and K-Ras isoforms, which are highly related and frequently mutated in cancer. Bisphenols are widespread in modern life because of their industrial application as plasticisers. Bisphenol A (BPA) is the best-known member and has gained significant scientific as well as public attention as an endocrine disrupting chemical, a fact that eventually led to its replacement. However, compounds used to replace BPA still contain the molecular scaffold of bisphenols. Here we show that BPA, BPAF, BPB, BPE, BPF, and an amine-substituted BPAF-derivate all interact with all GDP-bound Ras-Isoforms through binding to a common site on these proteins. NMR-, SOScat-, and GDI- assay-based data revealed a new bisphenol-induced, allosterically activated GDP-bound Ras conformation that define these plasticisers as Ras allosteric agonists.
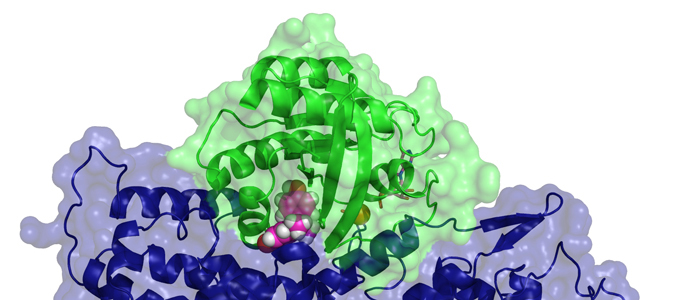
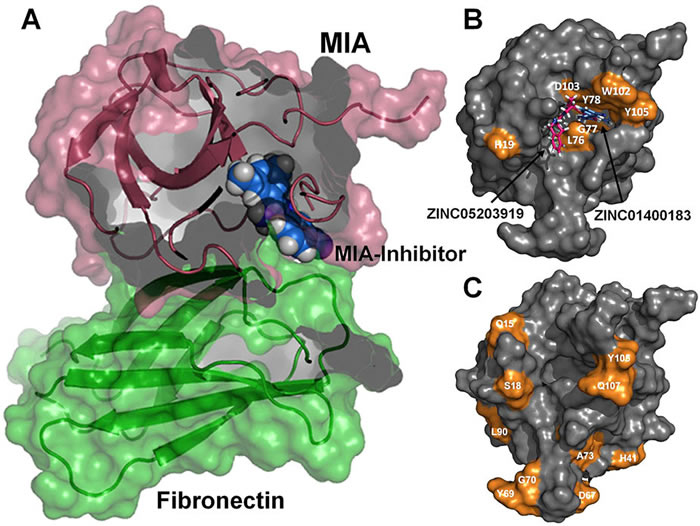
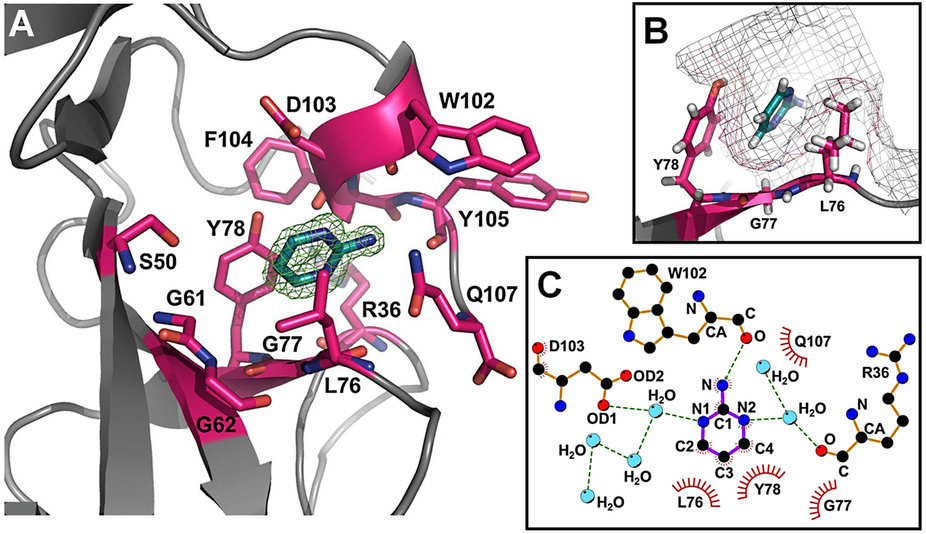 Melanoma inhibitory activity (MIA), an extracellular protein highly expressed by malignant melanoma cells, plays an important functional role in melanoma development, progression, and metastasis. After its secretion, MIA directly interacts with extracellular matrix proteins, such as fibronectin (FN). By this mechanism, MIA actively facilitates focal cell detachment from surrounding structures and strongly promotes tumour cell invasion and migration. Hence, the molecular understanding of MIA’s function provides a promising target for the development of new strategies in malignant melanoma therapy. Here, we describe for the first time the discovery of small molecules that are able to disrupt the MIA-FN complex by selectively binding to a new druggable pocket, which we could identify on MIA by structural analysis and fragment-based screening. Our findings may inspire novel drug discovery efforts aiming at a therapeutically effective treatment of melanoma by targeting MIA.
Melanoma inhibitory activity (MIA), an extracellular protein highly expressed by malignant melanoma cells, plays an important functional role in melanoma development, progression, and metastasis. After its secretion, MIA directly interacts with extracellular matrix proteins, such as fibronectin (FN). By this mechanism, MIA actively facilitates focal cell detachment from surrounding structures and strongly promotes tumour cell invasion and migration. Hence, the molecular understanding of MIA’s function provides a promising target for the development of new strategies in malignant melanoma therapy. Here, we describe for the first time the discovery of small molecules that are able to disrupt the MIA-FN complex by selectively binding to a new druggable pocket, which we could identify on MIA by structural analysis and fragment-based screening. Our findings may inspire novel drug discovery efforts aiming at a therapeutically effective treatment of melanoma by targeting MIA.
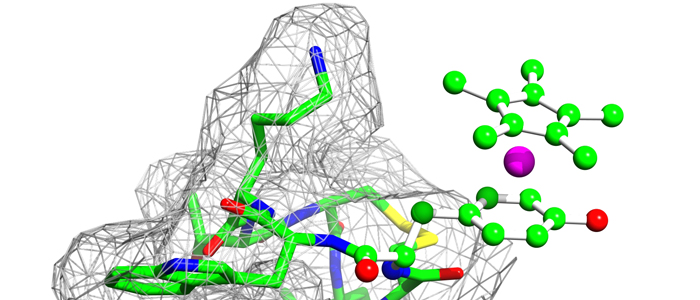
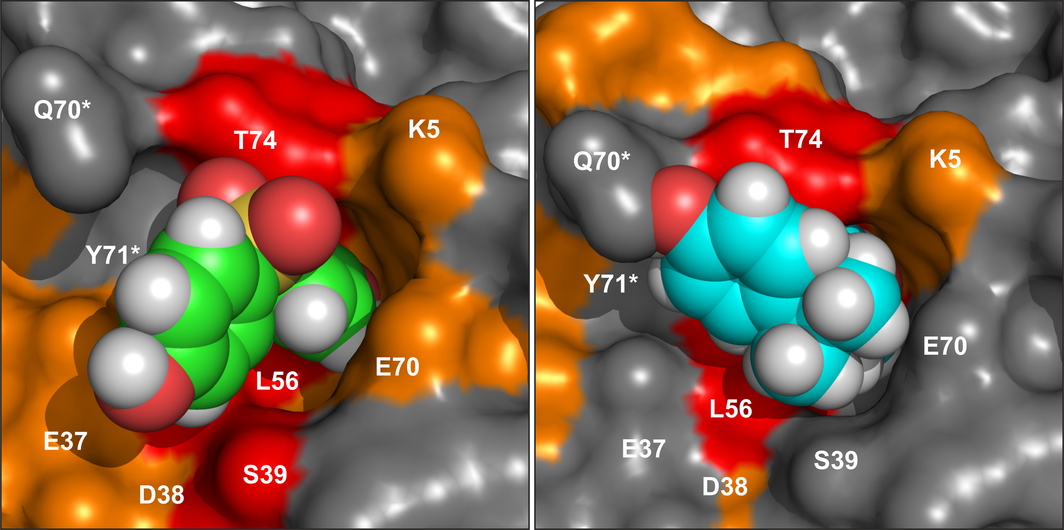 K-Ras4B is a small GTPase that belongs to the Ras superfamily of guanine nucleotide-binding proteins. GTPases function as molecular switches in cells and are key players in intracellular signalling. Ras has been identified as an oncogene and is mutated in more than 20% of human cancers. Here, we report that Bisphenol S binds into a binding pocket of K-Ras4B previously identified for various low molecular weight compounds. Our results advocate for more comprehensive safety studies on the toxicity of Bisphenol S, as it is frequently used for Bisphenol A-free food containers.
K-Ras4B is a small GTPase that belongs to the Ras superfamily of guanine nucleotide-binding proteins. GTPases function as molecular switches in cells and are key players in intracellular signalling. Ras has been identified as an oncogene and is mutated in more than 20% of human cancers. Here, we report that Bisphenol S binds into a binding pocket of K-Ras4B previously identified for various low molecular weight compounds. Our results advocate for more comprehensive safety studies on the toxicity of Bisphenol S, as it is frequently used for Bisphenol A-free food containers.
 Animal venoms, such as those from scorpions, are a potent source for new pharmacological substances. In this study we have determined the structure of the α-KTx3.8 (named as Bs6) scorpion toxin by multidimensional 1H homonuclear NMR spectroscopy and investigated its function by molecular dynamics (MD) simulations and electrophysiological measurements. Bs6 is a potent inhibitor of the Kv1.3 channel which plays an important role during the activation and proliferation of memory T-cells (TEM), which play an important role in autoimmune diseases.
Therefore, it could be an interesting target for treatment of autoimmune diseases. In this study, Bs6 was synthesised by solid phase synthesis and its three-dimensional (3D) structure has been determined. To gain a deeper insight into the interaction of Bs6 with different potassium channels like hKv1.1 and hKv1.3, the protein-protein complex was modelled based on known toxin-channel structures and tested for stability in MD simulations using GROMACS. The toxin-channel interaction was further analysed by electrophysiological measurements of different potassium channels like hKv1.3 and hKv7.1. As potassium channel inhibitors could play an important role to overcome autoimmune diseases like multiple sclerosis and type-1 diabetes mellitus, our data contributes to the understanding of the molecular mechanism of action and will ultimately help to develop new potent inhibitors in future.
Animal venoms, such as those from scorpions, are a potent source for new pharmacological substances. In this study we have determined the structure of the α-KTx3.8 (named as Bs6) scorpion toxin by multidimensional 1H homonuclear NMR spectroscopy and investigated its function by molecular dynamics (MD) simulations and electrophysiological measurements. Bs6 is a potent inhibitor of the Kv1.3 channel which plays an important role during the activation and proliferation of memory T-cells (TEM), which play an important role in autoimmune diseases.
Therefore, it could be an interesting target for treatment of autoimmune diseases. In this study, Bs6 was synthesised by solid phase synthesis and its three-dimensional (3D) structure has been determined. To gain a deeper insight into the interaction of Bs6 with different potassium channels like hKv1.1 and hKv1.3, the protein-protein complex was modelled based on known toxin-channel structures and tested for stability in MD simulations using GROMACS. The toxin-channel interaction was further analysed by electrophysiological measurements of different potassium channels like hKv1.3 and hKv7.1. As potassium channel inhibitors could play an important role to overcome autoimmune diseases like multiple sclerosis and type-1 diabetes mellitus, our data contributes to the understanding of the molecular mechanism of action and will ultimately help to develop new potent inhibitors in future.
Nucleotide Exchange: Implications for GTPase-Selective Antagonists.
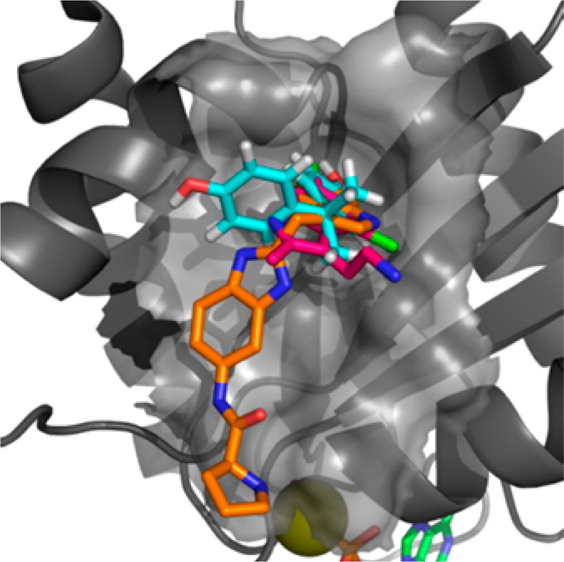 We show here for the first time that bisphenol A (10) has the capacity to interact directly with K-Ras and that Rheb weakly binds to bisphenol A (10) and 4,4′-biphenol derivatives. We have characterized these interactions at atomic resolution suggesting that these compounds sterically interfere with the Sos-mediated nucleotide exchange in H- and K-Ras. We show that 4,4′-biphenol (5) selectively inhibits Rheb signaling and induces cell death suggesting that this compound might be a novel candidate for treatment of tuberous sclerosis-mediated tumor growth. Our results propose a new mode of action for bisphenol A (10) that advocates a reduced exposure to this compound in our environment. Our data may lay the foundation for the future design of GTPaseselective antagonists with higher affinity to benefit of the treatment of cancer because KRas inhibition is regarded to be a promising strategy with a potential therapeutic window for targeting Sos in Ras-driven tumors.
We show here for the first time that bisphenol A (10) has the capacity to interact directly with K-Ras and that Rheb weakly binds to bisphenol A (10) and 4,4′-biphenol derivatives. We have characterized these interactions at atomic resolution suggesting that these compounds sterically interfere with the Sos-mediated nucleotide exchange in H- and K-Ras. We show that 4,4′-biphenol (5) selectively inhibits Rheb signaling and induces cell death suggesting that this compound might be a novel candidate for treatment of tuberous sclerosis-mediated tumor growth. Our results propose a new mode of action for bisphenol A (10) that advocates a reduced exposure to this compound in our environment. Our data may lay the foundation for the future design of GTPaseselective antagonists with higher affinity to benefit of the treatment of cancer because KRas inhibition is regarded to be a promising strategy with a potential therapeutic window for targeting Sos in Ras-driven tumors.
 Binding
of Leu-enkephalin and [RhIII(η5-Cp*)(η6-Tyr1)]Leu-enkephalin
to the recently published crystal structures of the μ- and δ-opioid
receptor is studied. Docking of free Leu-enkephalin reveals two
preferred conformations, one of which suggests an alternative binding
site for the tyrosine residue. Furthermore, the three-dimensional
solution structure of [RhIII(η5-Cp*)(η6-Tyr1)]Leu-enkephalin
was solved by using 2D NMR spectroscopic techniques.
Binding
of Leu-enkephalin and [RhIII(η5-Cp*)(η6-Tyr1)]Leu-enkephalin
to the recently published crystal structures of the μ- and δ-opioid
receptor is studied. Docking of free Leu-enkephalin reveals two
preferred conformations, one of which suggests an alternative binding
site for the tyrosine residue. Furthermore, the three-dimensional
solution structure of [RhIII(η5-Cp*)(η6-Tyr1)]Leu-enkephalin
was solved by using 2D NMR spectroscopic techniques.
 The bioconjugation of organometallic complexes with peptides has proven to be a novel approach for drug discovery. We report the facile and chemoselective reaction of tyrosine-containing G-protein-coupled receptor (GPCR) peptides with [Cp*Rh(H2O)3](OTf)2, in water, at room temperature, and at pH 5–6. We have focused on three important GPCR peptides; namely, [Tyr1]-leu-enkephalin, [Tyr4]-neurotensin(8-13), and [Tyr3]-octreotide, each of which has a different position for the tyrosine residue, together with competing functionalities. Importantly, all other functional groups present, i.e., amino, carboxyl, disulfide, phenyl, and indole, were not prominent sites of reactivity by the Cp*Rh tris aqua complex. Furthermore, the influence of the Cp*Rh moiety on the structure of [Tyr3]-octreotide was characterized by 2D NMR, resulting in the first representative structure of an organometallic-peptide complex. The biological consequences of these Cp*Rh-peptide complexes, with respect to GPCR binding and growth inhibition of MCF7 and HT29 cancer cells, will be presented for [(η6-Cp*Rh-Tyr1)-leu-enkephalin](OTf)2 and [(η6-Cp*Rh-Tyr3)-octreotide](OTf)2.
The bioconjugation of organometallic complexes with peptides has proven to be a novel approach for drug discovery. We report the facile and chemoselective reaction of tyrosine-containing G-protein-coupled receptor (GPCR) peptides with [Cp*Rh(H2O)3](OTf)2, in water, at room temperature, and at pH 5–6. We have focused on three important GPCR peptides; namely, [Tyr1]-leu-enkephalin, [Tyr4]-neurotensin(8-13), and [Tyr3]-octreotide, each of which has a different position for the tyrosine residue, together with competing functionalities. Importantly, all other functional groups present, i.e., amino, carboxyl, disulfide, phenyl, and indole, were not prominent sites of reactivity by the Cp*Rh tris aqua complex. Furthermore, the influence of the Cp*Rh moiety on the structure of [Tyr3]-octreotide was characterized by 2D NMR, resulting in the first representative structure of an organometallic-peptide complex. The biological consequences of these Cp*Rh-peptide complexes, with respect to GPCR binding and growth inhibition of MCF7 and HT29 cancer cells, will be presented for [(η6-Cp*Rh-Tyr1)-leu-enkephalin](OTf)2 and [(η6-Cp*Rh-Tyr3)-octreotide](OTf)2.
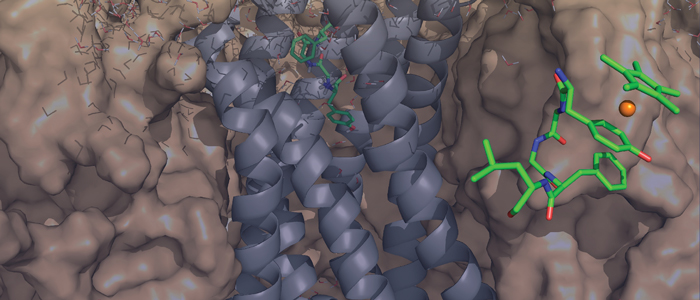
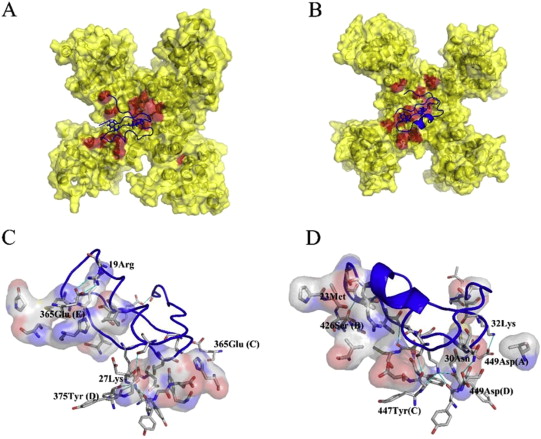
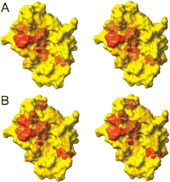 The oncoprotein MDM2 inhibits the tumor suppressor protein p53 by binding to the p53 transactivation domain. The p53 gene is inactivated in many human tumors either by mutations or by binding to oncogenic proteins. In some tumors, such as soft tissue sarcomas, overexpression of MDM2 inactivates an otherwise intact p53, disabling the genome integrity checkpoint and allowing cell cycle progression of defective cells. Disruption of the MDM2/p53 interaction leads to increased p53 levels and restored p53 transcriptional activity, indicating restoration of the genome integrity check and therapeutic potential for MDM2/p53 binding antagonists. Here, we show by multidimensional NMR spectroscopy that chalcones (1,3-diphenyl-2-propen-1-ones) are MDM2 inhibitors that bind to a subsite of the p53 binding cleft of human MDM2 (A and B). Biochemical experiments showed that these compounds can disrupt the MDM2/p53 protein complex, releasing p53 from both the p53/MDM2 and DNA-bound p53/MDM2 complexes. These results thus offer a starting basis for structure-based drug design of cancer therapeutics.
The oncoprotein MDM2 inhibits the tumor suppressor protein p53 by binding to the p53 transactivation domain. The p53 gene is inactivated in many human tumors either by mutations or by binding to oncogenic proteins. In some tumors, such as soft tissue sarcomas, overexpression of MDM2 inactivates an otherwise intact p53, disabling the genome integrity checkpoint and allowing cell cycle progression of defective cells. Disruption of the MDM2/p53 interaction leads to increased p53 levels and restored p53 transcriptional activity, indicating restoration of the genome integrity check and therapeutic potential for MDM2/p53 binding antagonists. Here, we show by multidimensional NMR spectroscopy that chalcones (1,3-diphenyl-2-propen-1-ones) are MDM2 inhibitors that bind to a subsite of the p53 binding cleft of human MDM2 (A and B). Biochemical experiments showed that these compounds can disrupt the MDM2/p53 protein complex, releasing p53 from both the p53/MDM2 and DNA-bound p53/MDM2 complexes. These results thus offer a starting basis for structure-based drug design of cancer therapeutics.