Fakultät für
Chemie
und Biochemie
Biochemie II
Biomolekulare NMR-Spektroskopie
Prof. Dr. Raphael Stoll
Gebäude NC 5/171
Universitätsstraße 150
D-44780 Bochum
Tel.: +49 234 32-25466
Fax: +49 234 32-05466
E-Mail: bionmr@rub.de







Signal Transduction
 "The nuclear factor of kappa light polypeptide gene enhancer in B-cells (NFκB) transcription factors play a critical role in human immune response. The family includes homodimers and heterodimers of five component proteins, which mediate different transcriptional responses and bind preferentially to different DNA sequences. Crystal structures of DNA complexes show that the dimers of the Rel-homology regions are structurally very similar. Differing DNA sequence preference together with structural similarity suggests that the dimers may differ in their dynamics. In this study, we present the first near-complete 15 N, 13 Cα/β , and HN backbone resonance assignments of two dimers of the dimerization domain (DD) of the NFκB1 (p50) protein (residues 241-351): the homodimer of two p50 domains and a heterodimer of the p50 DD with the p65 DD. As expected, the two dimers behave very similarly, with chemical shift differences between them largely concentrated in the dimer interface and attributable to specific differences in the amino acid sequences of p50 and p65. A comparison of the picosecond-nanosecond dynamics of the homo- and heterodimers also shows that the environment of p50 is similar, with an overall slightly reduced correlation time for the homodimer compared to the heterodimer, consistent with its slightly smaller molecular weight. These results demonstrate that NMR spectroscopy can be used to explore subtle changes in structure and dynamics that have the potential to give insights into differences in specificity that can be exploited in the design of new therapeutic agents.
"The nuclear factor of kappa light polypeptide gene enhancer in B-cells (NFκB) transcription factors play a critical role in human immune response. The family includes homodimers and heterodimers of five component proteins, which mediate different transcriptional responses and bind preferentially to different DNA sequences. Crystal structures of DNA complexes show that the dimers of the Rel-homology regions are structurally very similar. Differing DNA sequence preference together with structural similarity suggests that the dimers may differ in their dynamics. In this study, we present the first near-complete 15 N, 13 Cα/β , and HN backbone resonance assignments of two dimers of the dimerization domain (DD) of the NFκB1 (p50) protein (residues 241-351): the homodimer of two p50 domains and a heterodimer of the p50 DD with the p65 DD. As expected, the two dimers behave very similarly, with chemical shift differences between them largely concentrated in the dimer interface and attributable to specific differences in the amino acid sequences of p50 and p65. A comparison of the picosecond-nanosecond dynamics of the homo- and heterodimers also shows that the environment of p50 is similar, with an overall slightly reduced correlation time for the homodimer compared to the heterodimer, consistent with its slightly smaller molecular weight. These results demonstrate that NMR spectroscopy can be used to explore subtle changes in structure and dynamics that have the potential to give insights into differences in specificity that can be exploited in the design of new therapeutic agents.
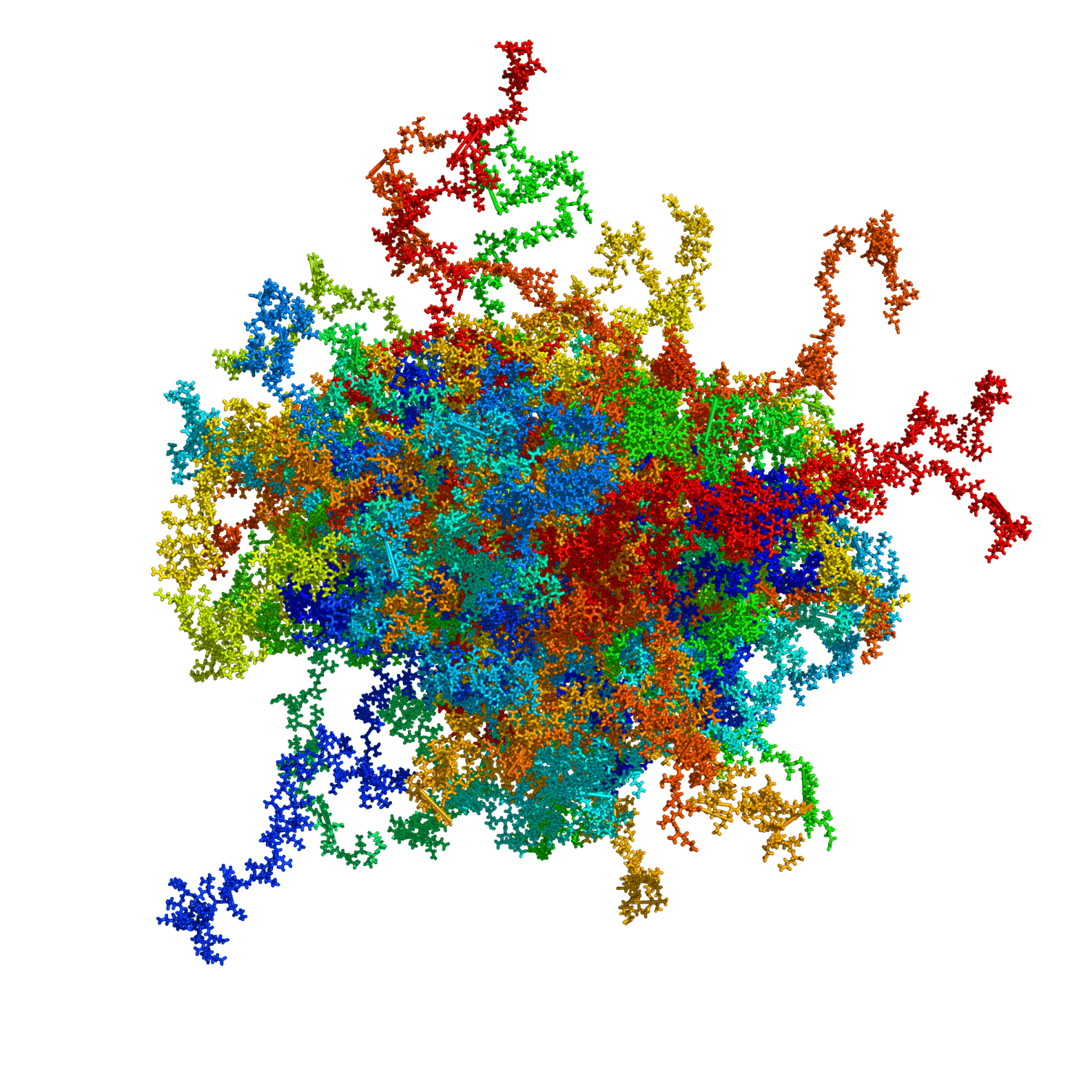 High Mobility Group Protein A1a (HMGA1a) is a highly abundant nuclear protein, which plays a crucial role during embryogenesis, cell differentiation, and neoplasia. Here, we present the first ever NMR-based structural ensemble of full length HMGA1a. Our results show that the protein is not completely random coil but adopts a compact structure consisting of transient long-range contacts, which is regulated by post-translational phosphorylation. The CK2-, cdc2- and cdc2/CK2-phosphorylated forms of HMGA1a each exhibit a different binding affinity towards the PRD2 element of the NFκB promoter. Our study identifies connected regions between phosphorylation sites in the wildtype ensemble that change considerably upon phosphorylation, indicating that these posttranslational modifications sites are part of an electrostatic contact network that alters the structural ensemble by shifting the conformational equilibrium. Moreover, ITC data reveal that the CK2-phosphorylated HMGA1a exhibits a different DNA promoter binding affinity for the PRD2 element. Furthermore, we present the first structural model for AT-hook 1 of HMGA1a that can adopt a transient α-helical structure, which might serve as an additional regulatory mechanism in HMAG1a. Our findings will help to develop new therapeutic strategies against HMGA1a-associated cancers by taking posttranslational modifications into consideration.
High Mobility Group Protein A1a (HMGA1a) is a highly abundant nuclear protein, which plays a crucial role during embryogenesis, cell differentiation, and neoplasia. Here, we present the first ever NMR-based structural ensemble of full length HMGA1a. Our results show that the protein is not completely random coil but adopts a compact structure consisting of transient long-range contacts, which is regulated by post-translational phosphorylation. The CK2-, cdc2- and cdc2/CK2-phosphorylated forms of HMGA1a each exhibit a different binding affinity towards the PRD2 element of the NFκB promoter. Our study identifies connected regions between phosphorylation sites in the wildtype ensemble that change considerably upon phosphorylation, indicating that these posttranslational modifications sites are part of an electrostatic contact network that alters the structural ensemble by shifting the conformational equilibrium. Moreover, ITC data reveal that the CK2-phosphorylated HMGA1a exhibits a different DNA promoter binding affinity for the PRD2 element. Furthermore, we present the first structural model for AT-hook 1 of HMGA1a that can adopt a transient α-helical structure, which might serve as an additional regulatory mechanism in HMAG1a. Our findings will help to develop new therapeutic strategies against HMGA1a-associated cancers by taking posttranslational modifications into consideration.
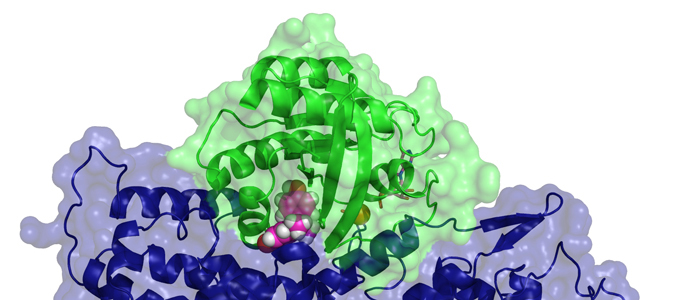
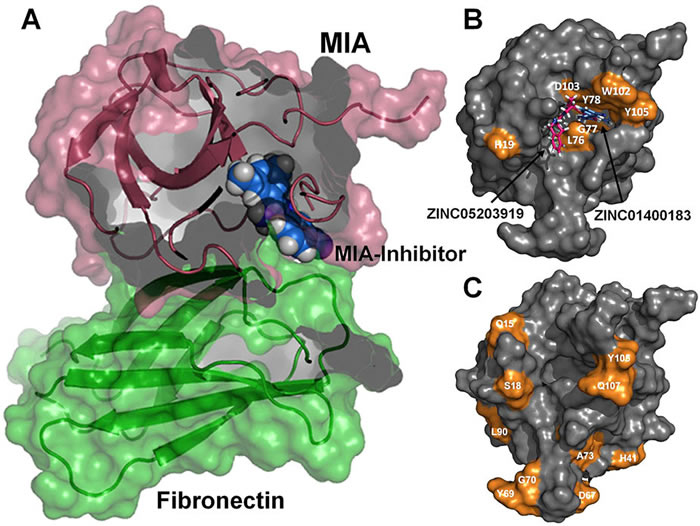
 Protein tyrosine phosphatase PTPN13, also known as PTP-BL in mice, is a large multi-domain non-transmembrane scaffolding protein with a molecular mass of 270 kDa. It is involved in the regulation of several cellular processes such as cytokinesis and actin-cytoskeletal rearrangement. The modular structure of PTPN13 consists of an N-terminal KIND domain, a FERM domain, and five PDZ domains, followed by a C-terminal protein tyrosine phosphatase domain. PDZ domains are among the most abundant protein modules and they play a crucial role in signal transduction of protein networks.
Protein tyrosine phosphatase PTPN13, also known as PTP-BL in mice, is a large multi-domain non-transmembrane scaffolding protein with a molecular mass of 270 kDa. It is involved in the regulation of several cellular processes such as cytokinesis and actin-cytoskeletal rearrangement. The modular structure of PTPN13 consists of an N-terminal KIND domain, a FERM domain, and five PDZ domains, followed by a C-terminal protein tyrosine phosphatase domain. PDZ domains are among the most abundant protein modules and they play a crucial role in signal transduction of protein networks.
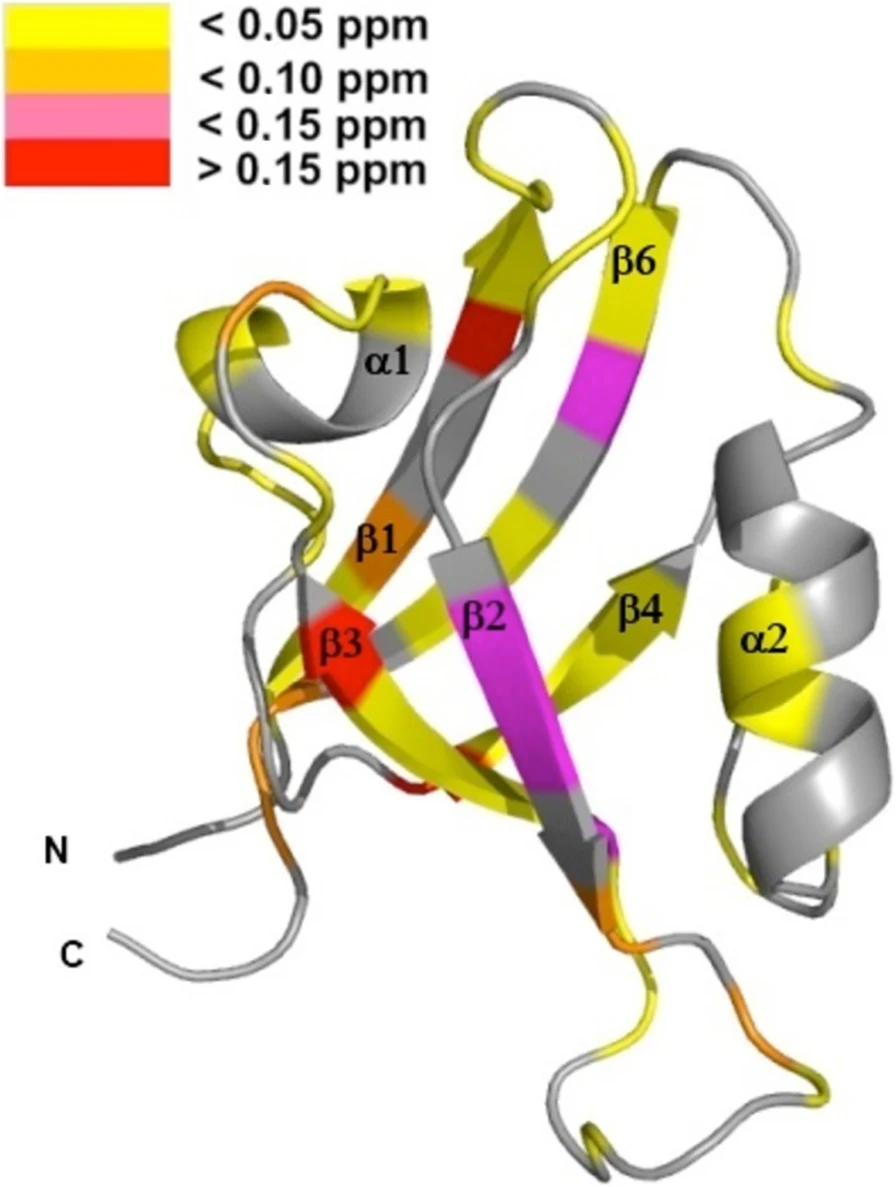
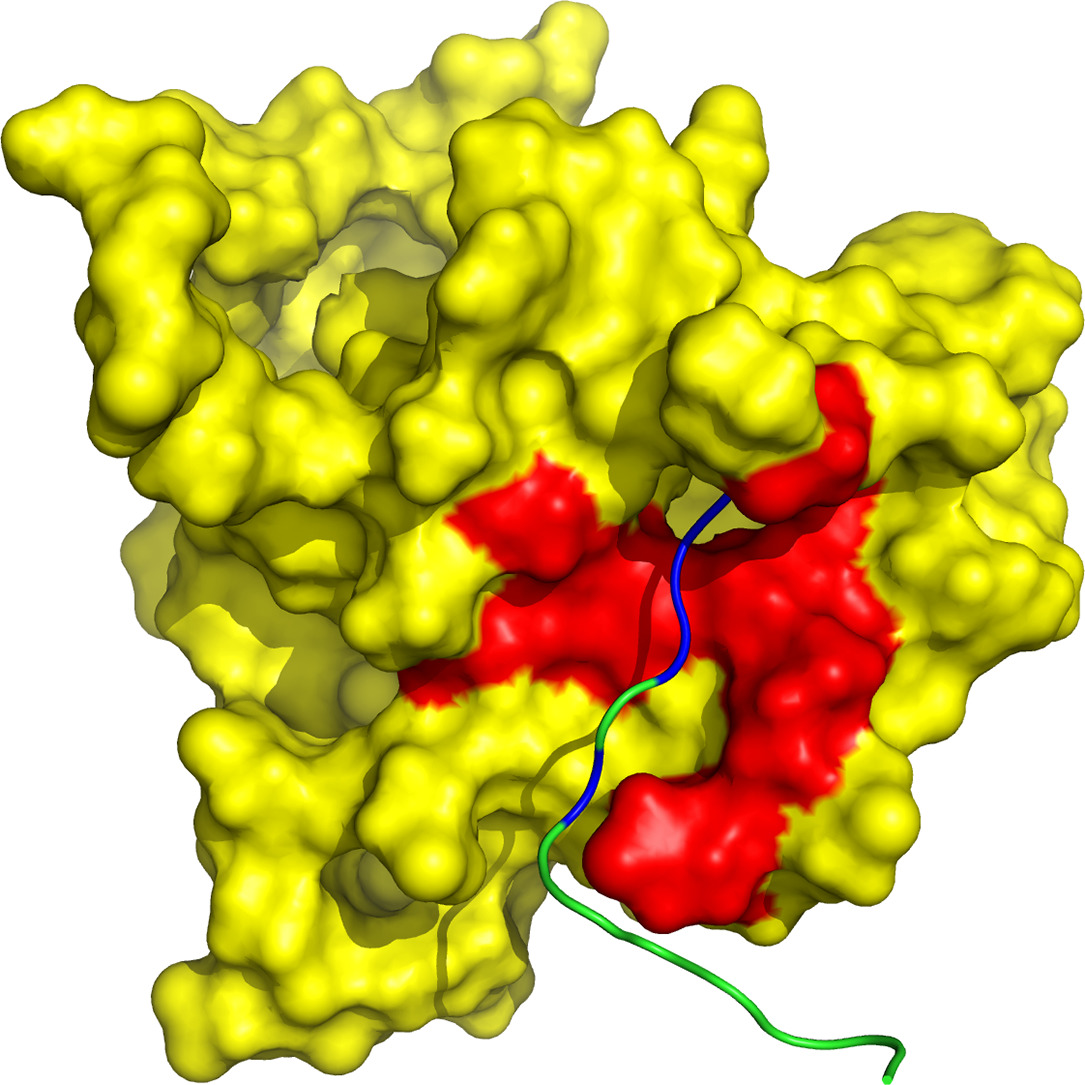 Protein tyrosine phosphatase PTPN13, also known as PTP-BL in mice, represents a large multi-domain non-transmembrane scaffolding protein that contains five consecutive PDZ domains. Here, we report the solution structures of the extended murine PTPN13 PDZ3 domain in its apo form and in complex with its physiological ligand, the carboxy-terminus of protein kinase C-related kinase-2 (PRK2), determined by multidimensional NMR spectroscopy. Both in its ligand-free state and when complexed to PRK2, PDZ3 of PTPN13 adopts the classical compact, globular D/E fold. PDZ3 of PTPN13 binds five carboxy-terminal amino acids of PRK2 via a groove located between the EB-strand and the DB-helix. The PRK2 peptide resides in the canonical PDZ3 binding cleft in an elongated manner and the amino acid side chains in position P0 and P-2, cysteine and aspartate, of the ligand face the groove between EB-strand and DB-helix, whereas the PRK2 side chains of tryptophan and alanine located in position P-1 and P-3 point away from the binding cleft. These structures are rare examples of selective class III ligand recognition by a PDZ domain and now provide a basis for the detailed structural investigation of the promiscuous interaction between the PDZ domains of PTPN13 and their ligands. They will also lead to a better understanding of the proposed scaffolding function of these domains in multi-protein complexes assembled by PTPN13 and could ultimately contribute to low molecular weight antagonists that might even act on the PRK2 signaling pathway to modulate rearrangements of the actin cytoskeleton.
Protein tyrosine phosphatase PTPN13, also known as PTP-BL in mice, represents a large multi-domain non-transmembrane scaffolding protein that contains five consecutive PDZ domains. Here, we report the solution structures of the extended murine PTPN13 PDZ3 domain in its apo form and in complex with its physiological ligand, the carboxy-terminus of protein kinase C-related kinase-2 (PRK2), determined by multidimensional NMR spectroscopy. Both in its ligand-free state and when complexed to PRK2, PDZ3 of PTPN13 adopts the classical compact, globular D/E fold. PDZ3 of PTPN13 binds five carboxy-terminal amino acids of PRK2 via a groove located between the EB-strand and the DB-helix. The PRK2 peptide resides in the canonical PDZ3 binding cleft in an elongated manner and the amino acid side chains in position P0 and P-2, cysteine and aspartate, of the ligand face the groove between EB-strand and DB-helix, whereas the PRK2 side chains of tryptophan and alanine located in position P-1 and P-3 point away from the binding cleft. These structures are rare examples of selective class III ligand recognition by a PDZ domain and now provide a basis for the detailed structural investigation of the promiscuous interaction between the PDZ domains of PTPN13 and their ligands. They will also lead to a better understanding of the proposed scaffolding function of these domains in multi-protein complexes assembled by PTPN13 and could ultimately contribute to low molecular weight antagonists that might even act on the PRK2 signaling pathway to modulate rearrangements of the actin cytoskeleton.
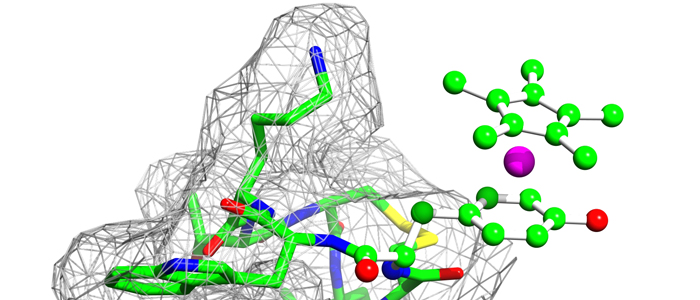
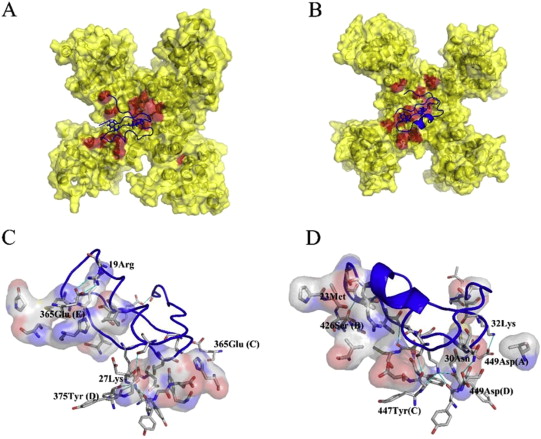
 The protein family of small GTPases controls cellular processes by acting as a binary switch between an active and an inactive state. The most prominent family members are H-Ras, N-Ras, and K-Ras isoforms, which are highly related and frequently mutated in cancer. Bisphenols are widespread in modern life because of their industrial application as plasticisers. Bisphenol A (BPA) is the best-known member and has gained significant scientific as well as public attention as an endocrine disrupting chemical, a fact that eventually led to its replacement. However, compounds used to replace BPA still contain the molecular scaffold of bisphenols. Here we show that BPA, BPAF, BPB, BPE, BPF, and an amine-substituted BPAF-derivate all interact with all GDP-bound Ras-Isoforms through binding to a common site on these proteins. NMR-, SOScat-, and GDI- assay-based data revealed a new bisphenol-induced, allosterically activated GDP-bound Ras conformation that define these plasticisers as Ras allosteric agonists.
The protein family of small GTPases controls cellular processes by acting as a binary switch between an active and an inactive state. The most prominent family members are H-Ras, N-Ras, and K-Ras isoforms, which are highly related and frequently mutated in cancer. Bisphenols are widespread in modern life because of their industrial application as plasticisers. Bisphenol A (BPA) is the best-known member and has gained significant scientific as well as public attention as an endocrine disrupting chemical, a fact that eventually led to its replacement. However, compounds used to replace BPA still contain the molecular scaffold of bisphenols. Here we show that BPA, BPAF, BPB, BPE, BPF, and an amine-substituted BPAF-derivate all interact with all GDP-bound Ras-Isoforms through binding to a common site on these proteins. NMR-, SOScat-, and GDI- assay-based data revealed a new bisphenol-induced, allosterically activated GDP-bound Ras conformation that define these plasticisers as Ras allosteric agonists.
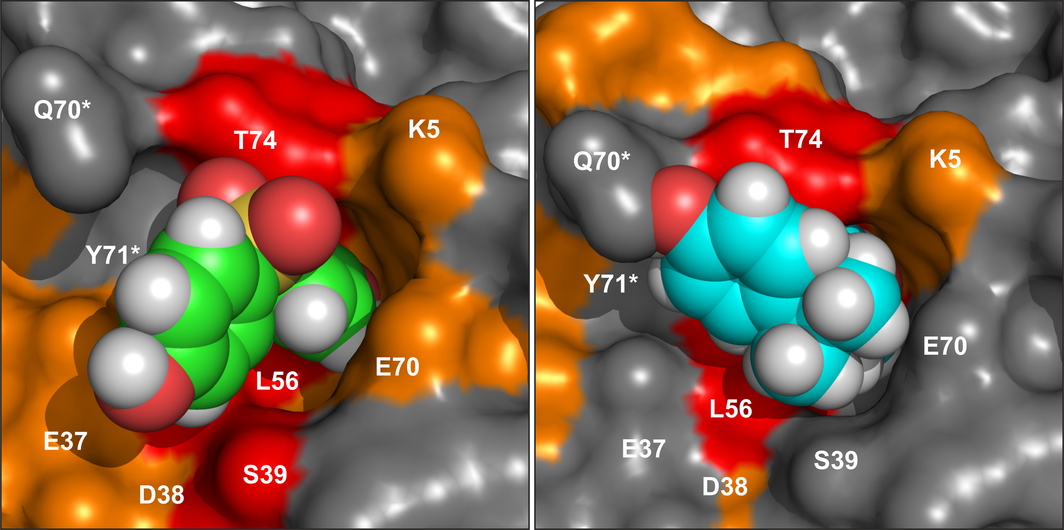 K-Ras4B is a small GTPase that belongs to the Ras superfamily of guanine nucleotide-binding proteins. GTPases function as molecular switches in cells and are key players in intracellular signalling. Ras has been identified as an oncogene and is mutated in more than 20% of human cancers. Here, we report that Bisphenol S binds into a binding pocket of K-Ras4B previously identified for various low molecular weight compounds. Our results advocate for more comprehensive safety studies on the toxicity of Bisphenol S, as it is frequently used for Bisphenol A-free food containers.
K-Ras4B is a small GTPase that belongs to the Ras superfamily of guanine nucleotide-binding proteins. GTPases function as molecular switches in cells and are key players in intracellular signalling. Ras has been identified as an oncogene and is mutated in more than 20% of human cancers. Here, we report that Bisphenol S binds into a binding pocket of K-Ras4B previously identified for various low molecular weight compounds. Our results advocate for more comprehensive safety studies on the toxicity of Bisphenol S, as it is frequently used for Bisphenol A-free food containers.
 Animal venoms, such as those from scorpions, are a potent source for new pharmacological substances. In this study we have determined the structure of the α-KTx3.8 (named as Bs6) scorpion toxin by multidimensional 1H homonuclear NMR spectroscopy and investigated its function by molecular dynamics (MD) simulations and electrophysiological measurements. Bs6 is a potent inhibitor of the Kv1.3 channel which plays an important role during the activation and proliferation of memory T-cells (TEM), which play an important role in autoimmune diseases.
Therefore, it could be an interesting target for treatment of autoimmune diseases. In this study, Bs6 was synthesised by solid phase synthesis and its three-dimensional (3D) structure has been determined. To gain a deeper insight into the interaction of Bs6 with different potassium channels like hKv1.1 and hKv1.3, the protein-protein complex was modelled based on known toxin-channel structures and tested for stability in MD simulations using GROMACS. The toxin-channel interaction was further analysed by electrophysiological measurements of different potassium channels like hKv1.3 and hKv7.1. As potassium channel inhibitors could play an important role to overcome autoimmune diseases like multiple sclerosis and type-1 diabetes mellitus, our data contributes to the understanding of the molecular mechanism of action and will ultimately help to develop new potent inhibitors in future.
Animal venoms, such as those from scorpions, are a potent source for new pharmacological substances. In this study we have determined the structure of the α-KTx3.8 (named as Bs6) scorpion toxin by multidimensional 1H homonuclear NMR spectroscopy and investigated its function by molecular dynamics (MD) simulations and electrophysiological measurements. Bs6 is a potent inhibitor of the Kv1.3 channel which plays an important role during the activation and proliferation of memory T-cells (TEM), which play an important role in autoimmune diseases.
Therefore, it could be an interesting target for treatment of autoimmune diseases. In this study, Bs6 was synthesised by solid phase synthesis and its three-dimensional (3D) structure has been determined. To gain a deeper insight into the interaction of Bs6 with different potassium channels like hKv1.1 and hKv1.3, the protein-protein complex was modelled based on known toxin-channel structures and tested for stability in MD simulations using GROMACS. The toxin-channel interaction was further analysed by electrophysiological measurements of different potassium channels like hKv1.3 and hKv7.1. As potassium channel inhibitors could play an important role to overcome autoimmune diseases like multiple sclerosis and type-1 diabetes mellitus, our data contributes to the understanding of the molecular mechanism of action and will ultimately help to develop new potent inhibitors in future.
Nucleotide Exchange: Implications for GTPase-Selective Antagonists.
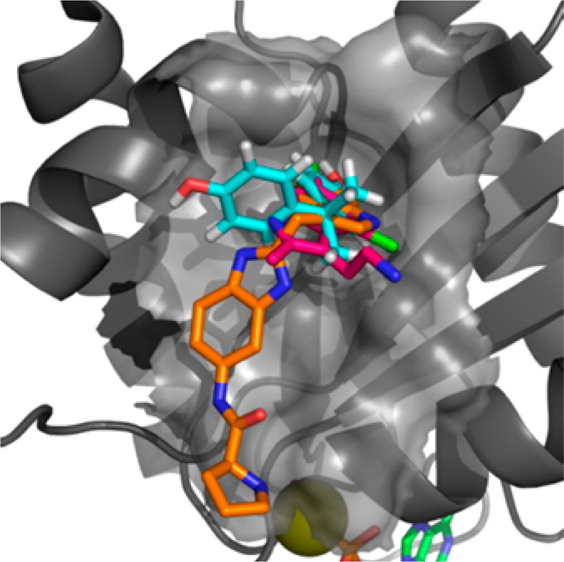 We show here for the first time that bisphenol A (10) has the capacity to interact directly with K-Ras and that Rheb weakly binds to bisphenol A (10) and 4,4′-biphenol derivatives. We have characterized these interactions at atomic resolution suggesting that these compounds sterically interfere with the Sos-mediated nucleotide exchange in H- and K-Ras. We show that 4,4′-biphenol (5) selectively inhibits Rheb signaling and induces cell death suggesting that this compound might be a novel candidate for treatment of tuberous sclerosis-mediated tumor growth. Our results propose a new mode of action for bisphenol A (10) that advocates a reduced exposure to this compound in our environment. Our data may lay the foundation for the future design of GTPaseselective antagonists with higher affinity to benefit of the treatment of cancer because KRas inhibition is regarded to be a promising strategy with a potential therapeutic window for targeting Sos in Ras-driven tumors.
We show here for the first time that bisphenol A (10) has the capacity to interact directly with K-Ras and that Rheb weakly binds to bisphenol A (10) and 4,4′-biphenol derivatives. We have characterized these interactions at atomic resolution suggesting that these compounds sterically interfere with the Sos-mediated nucleotide exchange in H- and K-Ras. We show that 4,4′-biphenol (5) selectively inhibits Rheb signaling and induces cell death suggesting that this compound might be a novel candidate for treatment of tuberous sclerosis-mediated tumor growth. Our results propose a new mode of action for bisphenol A (10) that advocates a reduced exposure to this compound in our environment. Our data may lay the foundation for the future design of GTPaseselective antagonists with higher affinity to benefit of the treatment of cancer because KRas inhibition is regarded to be a promising strategy with a potential therapeutic window for targeting Sos in Ras-driven tumors.
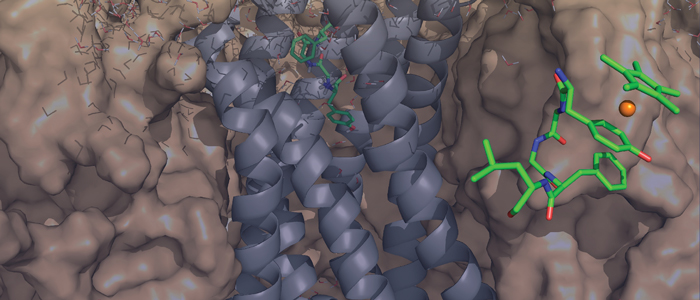
 Binding
of Leu-enkephalin and [RhIII(η5-Cp*)(η6-Tyr1)]Leu-enkephalin
to the recently published crystal structures of the μ- and δ-opioid
receptor is studied. Docking of free Leu-enkephalin reveals two
preferred conformations, one of which suggests an alternative binding
site for the tyrosine residue. Furthermore, the three-dimensional
solution structure of [RhIII(η5-Cp*)(η6-Tyr1)]Leu-enkephalin
was solved by using 2D NMR spectroscopic techniques.
Binding
of Leu-enkephalin and [RhIII(η5-Cp*)(η6-Tyr1)]Leu-enkephalin
to the recently published crystal structures of the μ- and δ-opioid
receptor is studied. Docking of free Leu-enkephalin reveals two
preferred conformations, one of which suggests an alternative binding
site for the tyrosine residue. Furthermore, the three-dimensional
solution structure of [RhIII(η5-Cp*)(η6-Tyr1)]Leu-enkephalin
was solved by using 2D NMR spectroscopic techniques.
 The bioconjugation of organometallic complexes with peptides has proven to be a novel approach for drug discovery. We report the facile and chemoselective reaction of tyrosine-containing G-protein-coupled receptor (GPCR) peptides with [Cp*Rh(H2O)3](OTf)2, in water, at room temperature, and at pH 5–6. We have focused on three important GPCR peptides; namely, [Tyr1]-leu-enkephalin, [Tyr4]-neurotensin(8-13), and [Tyr3]-octreotide, each of which has a different position for the tyrosine residue, together with competing functionalities. Importantly, all other functional groups present, i.e., amino, carboxyl, disulfide, phenyl, and indole, were not prominent sites of reactivity by the Cp*Rh tris aqua complex. Furthermore, the influence of the Cp*Rh moiety on the structure of [Tyr3]-octreotide was characterized by 2D NMR, resulting in the first representative structure of an organometallic-peptide complex. The biological consequences of these Cp*Rh-peptide complexes, with respect to GPCR binding and growth inhibition of MCF7 and HT29 cancer cells, will be presented for [(η6-Cp*Rh-Tyr1)-leu-enkephalin](OTf)2 and [(η6-Cp*Rh-Tyr3)-octreotide](OTf)2.
The bioconjugation of organometallic complexes with peptides has proven to be a novel approach for drug discovery. We report the facile and chemoselective reaction of tyrosine-containing G-protein-coupled receptor (GPCR) peptides with [Cp*Rh(H2O)3](OTf)2, in water, at room temperature, and at pH 5–6. We have focused on three important GPCR peptides; namely, [Tyr1]-leu-enkephalin, [Tyr4]-neurotensin(8-13), and [Tyr3]-octreotide, each of which has a different position for the tyrosine residue, together with competing functionalities. Importantly, all other functional groups present, i.e., amino, carboxyl, disulfide, phenyl, and indole, were not prominent sites of reactivity by the Cp*Rh tris aqua complex. Furthermore, the influence of the Cp*Rh moiety on the structure of [Tyr3]-octreotide was characterized by 2D NMR, resulting in the first representative structure of an organometallic-peptide complex. The biological consequences of these Cp*Rh-peptide complexes, with respect to GPCR binding and growth inhibition of MCF7 and HT29 cancer cells, will be presented for [(η6-Cp*Rh-Tyr1)-leu-enkephalin](OTf)2 and [(η6-Cp*Rh-Tyr3)-octreotide](OTf)2.
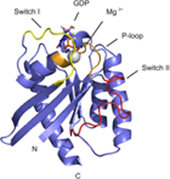 Rheb is a homolog of Ras GTPase that regulates cell growth, prolife
ration, and regeneration via mammalian target of rapamycin (mTOR). Because of the well established potential of activated Ras to promote survival, we sought to investigate the ability of Rheb signaling to phenocopy Ras. We found that overexpression of lipid-anchored Rheb enhanced the apoptotic effects induced by UV light, TNFα, or tunicamycin in an mTOR complex 1 (mTORC1)-dependent manner. Knocking down endogenous Rheb or applying rapamycin led to partial protection, identifying Rheb as a mediator of cell death. Ras and c-Raf kinase opposed the apoptotic effects induced by UV light or TNFα but did not prevent Rheb-mediated apoptosis. To gain structural insight into the signaling mechanisms, we determined the structure of Rheb-GDP by NMR. The complex adopts the typical canonical fold of RasGTPases and displays the characteristic GDP-dependent picosecond to nanosecond backbone dynamics of the switch I and switch II regions. NMR revealed Ras effector-like binding of activated Rheb to the c-Raf-Ras-binding domain (RBD), but the affinity was 1000-fold lower than the Ras/RBD interaction, suggesting a lack of functional interaction. shRNA-mediated knockdown of apoptosis signal-regulating kinase 1 (ASK-1) strongly reduced UV or TNFα-induced apoptosis and suppressed enhancement by Rheb overexpression. In conclusion, Rheb-mTOR activation not only promotes normal cell growth but also enhances apoptosis in response to diverse toxic stimuli via an ASK-1-mediated mechanism. Pharmacological regulation of the Rheb/mTORC1 pathway using rapamycin should take the presence of cellular stress into consideration, as this may have clinical implications.
Rheb is a homolog of Ras GTPase that regulates cell growth, prolife
ration, and regeneration via mammalian target of rapamycin (mTOR). Because of the well established potential of activated Ras to promote survival, we sought to investigate the ability of Rheb signaling to phenocopy Ras. We found that overexpression of lipid-anchored Rheb enhanced the apoptotic effects induced by UV light, TNFα, or tunicamycin in an mTOR complex 1 (mTORC1)-dependent manner. Knocking down endogenous Rheb or applying rapamycin led to partial protection, identifying Rheb as a mediator of cell death. Ras and c-Raf kinase opposed the apoptotic effects induced by UV light or TNFα but did not prevent Rheb-mediated apoptosis. To gain structural insight into the signaling mechanisms, we determined the structure of Rheb-GDP by NMR. The complex adopts the typical canonical fold of RasGTPases and displays the characteristic GDP-dependent picosecond to nanosecond backbone dynamics of the switch I and switch II regions. NMR revealed Ras effector-like binding of activated Rheb to the c-Raf-Ras-binding domain (RBD), but the affinity was 1000-fold lower than the Ras/RBD interaction, suggesting a lack of functional interaction. shRNA-mediated knockdown of apoptosis signal-regulating kinase 1 (ASK-1) strongly reduced UV or TNFα-induced apoptosis and suppressed enhancement by Rheb overexpression. In conclusion, Rheb-mTOR activation not only promotes normal cell growth but also enhances apoptosis in response to diverse toxic stimuli via an ASK-1-mediated mechanism. Pharmacological regulation of the Rheb/mTORC1 pathway using rapamycin should take the presence of cellular stress into consideration, as this may have clinical implications.
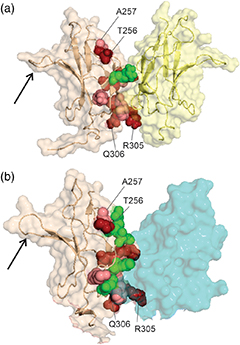
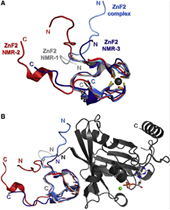 Nucleoporin (Nup) 153 is a highly mobile, multifunctional, and essential nuclear pore protein. It contains four zinc finger motifs that are thought to be crucial for the regulation of transportreceptor/ cargo interactions via their binding to the small guanine nucleotide binding protein, Ran. We found this interaction to be independent of the phosphorylation state of the nucleotide. Ran binds with the highest affinity to the second zinc finger motif of Nup153 (Nup153ZnF2). Here we present the crystal structure of this complex (B), revealing a new type of Ran-Ran interaction partner interface together with the solution structure of Nup153ZnF2 (A). According to our complex structure, Nup153ZnF2 binding to Ran excludes the formation of a Ran-importin-b complex. This finding suggests a local Nup153-mediated Ran reservoir at the nucleoplasmic distal ring of the nuclear pore, where nucleotide exchange may take place in a ternary Nup153-Ran-RCC1 complex, so that import complexes are efficiently terminated.
Nucleoporin (Nup) 153 is a highly mobile, multifunctional, and essential nuclear pore protein. It contains four zinc finger motifs that are thought to be crucial for the regulation of transportreceptor/ cargo interactions via their binding to the small guanine nucleotide binding protein, Ran. We found this interaction to be independent of the phosphorylation state of the nucleotide. Ran binds with the highest affinity to the second zinc finger motif of Nup153 (Nup153ZnF2). Here we present the crystal structure of this complex (B), revealing a new type of Ran-Ran interaction partner interface together with the solution structure of Nup153ZnF2 (A). According to our complex structure, Nup153ZnF2 binding to Ran excludes the formation of a Ran-importin-b complex. This finding suggests a local Nup153-mediated Ran reservoir at the nucleoplasmic distal ring of the nuclear pore, where nucleotide exchange may take place in a ternary Nup153-Ran-RCC1 complex, so that import complexes are efficiently terminated.
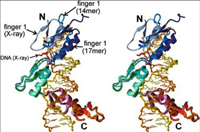 The zinc finger domain of the Wilms tumor suppressor protein (WT1) contains four canonical Cys2His2 zinc fingers. WT1 binds preferentially to DNA sequences that are closely related to the EGR-1 consensus site. We report the structure determination by both X-ray crystallography and NMR spectroscopy of the WT1 zinc finger domain in complex with DNA. The X-ray structure was determined for the complex with a cognate 14 base-pair oligonucleotide, and composite X-ray/NMR structures were determined for complexes with both the 14 base-pair and an extended 17 base-pair DNA. This combined approach allowed unambiguous determination of the position of the first zinc finger, which is influenced by lattice contacts in the crystal structure. The crystal structure shows the second, third and fourth zinc finger domains inserted deep into the major groove of the DNA where they make base-specific interactions. The DNA duplex is distorted in the vicinity of the first zinc finger, with a cytidine twisted and tilted out of the base stack to pack against finger 1 and the tip of finger 2. By contrast, the composite X-ray/NMR structures show that finger 1 continues to follow the major groove in the solution complexes. However, the orientation of the helix is non-canonical, and the fingertip and the N terminus of the helix project out of the major groove; as a consequence, the zinc finger side-chains that are commonly involved in base recognition make no contact with the DNA. We conclude that finger 1 helps to anchor WT1 to the DNA by amplifying the binding affinity although it does not contribute significantly to binding specificity. The structures provide molecular level insights into the potential consequences of mutations in zinc fingers 2 and 3 that are associated with Denys-Drash syndrome and nephritic syndrome. The mutations are of two types, and either destabilize the zinc finger structure or replace key base contact residues.
The zinc finger domain of the Wilms tumor suppressor protein (WT1) contains four canonical Cys2His2 zinc fingers. WT1 binds preferentially to DNA sequences that are closely related to the EGR-1 consensus site. We report the structure determination by both X-ray crystallography and NMR spectroscopy of the WT1 zinc finger domain in complex with DNA. The X-ray structure was determined for the complex with a cognate 14 base-pair oligonucleotide, and composite X-ray/NMR structures were determined for complexes with both the 14 base-pair and an extended 17 base-pair DNA. This combined approach allowed unambiguous determination of the position of the first zinc finger, which is influenced by lattice contacts in the crystal structure. The crystal structure shows the second, third and fourth zinc finger domains inserted deep into the major groove of the DNA where they make base-specific interactions. The DNA duplex is distorted in the vicinity of the first zinc finger, with a cytidine twisted and tilted out of the base stack to pack against finger 1 and the tip of finger 2. By contrast, the composite X-ray/NMR structures show that finger 1 continues to follow the major groove in the solution complexes. However, the orientation of the helix is non-canonical, and the fingertip and the N terminus of the helix project out of the major groove; as a consequence, the zinc finger side-chains that are commonly involved in base recognition make no contact with the DNA. We conclude that finger 1 helps to anchor WT1 to the DNA by amplifying the binding affinity although it does not contribute significantly to binding specificity. The structures provide molecular level insights into the potential consequences of mutations in zinc fingers 2 and 3 that are associated with Denys-Drash syndrome and nephritic syndrome. The mutations are of two types, and either destabilize the zinc finger structure or replace key base contact residues.
 The conformation of thymosin beta 9 in solution of 40% (v/v) 1,1,1,3,3,3-hexafluoro-2-propanol-d2 in water has been investigated by two-dimensional 1H-nmr spectroscopy. Under this condition thymosin beta 9 adopts an ordered structure. The determination of the conformation of the peptide was based on a set of 304 approximate interproton distance constraints derived from nuclear Overhauser enhancement measurements. The conformation of thymosin beta 9 includes two helical regions from residues 4 to 27 and 32 to 41. The two helices are separated by a poorly defined loop region between amino acids 28 and 31; the N-terminus of thymosin beta 9 shows random-coil structure only.
The conformation of thymosin beta 9 in solution of 40% (v/v) 1,1,1,3,3,3-hexafluoro-2-propanol-d2 in water has been investigated by two-dimensional 1H-nmr spectroscopy. Under this condition thymosin beta 9 adopts an ordered structure. The determination of the conformation of the peptide was based on a set of 304 approximate interproton distance constraints derived from nuclear Overhauser enhancement measurements. The conformation of thymosin beta 9 includes two helical regions from residues 4 to 27 and 32 to 41. The two helices are separated by a poorly defined loop region between amino acids 28 and 31; the N-terminus of thymosin beta 9 shows random-coil structure only.