HUMANGENETIK
Huntington Zentrum NRW
Information
Ein besonderes Interessensgebiet der Humangenetik an der Ruhr-Universität Bochum ist die Huntington-Erkrankung (Morbus Huntington). Seit der Entdeckung der ursächlichen (CAG)n-Verlängerung 1993 wird neben genetischer Beratung und Betreuung von Risikopersonen, Patienten und Familienangehörigen die molekulargenetische Testung für M. Huntington angeboten. In der besonderen Bochumer Konstellation (Diagnostik und Beratung in der Humangenetik; klinische Versorgung auf der Huntington-Station, Ansprechpartner PD Dr. med. Carsten Saft sowie psychologische und psychosoziale Betreuung in der Neurologie des St. Josef-Hospitals; psychiatrische Versorgung im Westfälischen Zentrum für Psychiatrie) wurde im Jahre 1993 das Huntington Zentrum NRW (HZ-NZW) gegründet. Dieses Zentrum gewährleistet durch intensive Interaktion der Humangenetiker und Ärzte mit besonderer klinischer Kompetenz umfassende psychosoziale Betreuung nicht nur für direkt Betroffene sondern auch für deren Umfeld.
Morbus Huntington
M. Huntington ist eine vererbte neurodegenerative Erkrankung, die normalerweise im mittleren bzw. späten Lebensalter durch fortschreitende Bewegungsstörungen sowie kognitive und psychiatrische Symptome gekennzeichnet ist. Von den Betroffenen selbst werden oft die Bewegungsstörungen als erstes wahrgenommen. Diese sind gekennzeichnet durch unwillkürliche, plötzliche, rasche, unregelmäßige und nicht vorhersehbare Bewegungen der Extremitäten, des Gesichts, des Halses und des Rumpfes. Dem vorausgehend, fallen der Familie und dem Umfeld des Betroffenen jedoch häufig Persönlichkeitsveränderungen des Betroffenen auf.
M. Huntington kommt weltweit vor, wobei die Krankheitshäufigkeit stark variiert und am häufigsten unter Menschen mit nordeuropäischer Abstammung zu finden ist. Die Prävalenz (Krankheitshäufigkeit) in der westlichen Hemisphäre wird mit 7-10 auf 100 000 Einwohner angegeben. Wie bereits oben erwähnt liegt das mittlere Erkrankungsalter liegt bei 40 Jahren, wobei auch juvenile (<20 Jahre) und späte (>70 Jahre) Manifestationen der Erkrankung vorkommen können.
Obwohl M. Huntington vergleichsweise selten anzutreffen ist, hat eine Diagnose weitreichende Folgen für die Betroffenen und deren Familien. Verwandte eines Betroffenen, für die ein Risiko besteht die Mutation ebenfalls zu tragen, stehen oft vor der schwierigen Entscheidung der prädiktiven genetischen Testung. Die folgenden Abschnitte geben nähere Informationen zur Huntingtin-Mutation, Erbang und genetischer Testung.
Wie sieht die Mutation im Huntingtin-Gen aus?
Die 1993 entdeckte Ursache von M. Huntington liegt in einer Mutation des Huntingtin-Gens (HTT), die in einer übermäßigen Wiederholung von drei Basen (CAG) im Exon 1 des HTT-Gens auf Chromosom 4 besteht (s. Abb. 1). Die Basenwiederholung führt zu einer überlangen, ununterbrochenen Kette ein und derselben Aminosäure (Glutamin), wodurch die normale Funktion des Huntingtin-Proteins gestört wird.
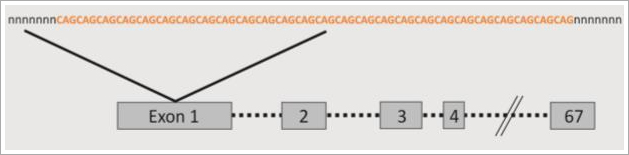
Abb. 1 CAG-Wiederholungen im Huntingtin-Gen (HTT). Im Exon 1 des Gens findet sich das Wiederholungsmotiv (CAG)n. Eine übermäßige Wiederholung der drei Basen führt zu M. Huntington.
Wie wird Morbus Huntington vererbt?
Jeder Mensch besitzt zwei Kopien des Huntingtin-Gens - eine wurde von der Mutter vererbt, die andere vom Vater. M. Huntington wird autosomal dominant vererbt, was bedeutet, dass es nur einer verlängerten Kopie bedarf, um die Krankheit zu verursachen. Die nicht-verlängerte Kopie des Gens ist nicht in der Lage die fehlende oder veränderte Funktion der mutierten Kopie aufzufangen.
Trägt ein Elternteil eine verlängerte Huntingtin-Kopie, wird diese an die Hälfte der Nachkommen weitervererbt, somit liegt das Wiederholungsrisiko bei 50% (Abb. 2). Da das Huntingtin-Gen auf einem Autosom liegt, also nicht auf einem der Geschlechtschromosomen X oder Y, tritt die Erkrankung in beiden Geschlechtern auf. Männer und Frauen sind also in gleicher Weise betroffen. Die Erkrankung überspringt keine Generationen, bei Nachkommen gesunder Familienmitglieder mit zwei kurzen Kopien des Huntingtin-Gens tritt die Erkrankung nicht auf.
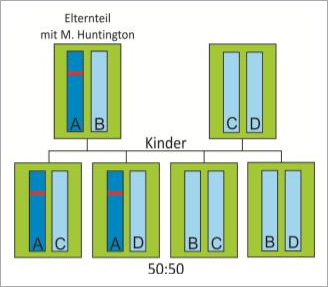
Abb. 2 Autosomal dominante Vererbung von M. Huntington. Trägt ein Elternteil die Huntingtin-Mutation, wird diese an die Hälfte der Nachkommen weitervererbt, das Wiederholungsrisiko liegt somit bei 50%.
Welche CAG-Länge führt zu M. Huntington?
Die meisten Menschen tragen zwei “normale” CAG-Längen im Huntingtin-Gen. Normalkopien umfassen bis zu 35 CAG-Wiederholungen, wobei am häufigsten Längen um 17 CAG zu beobachten sind (Abb. 3). Die meisten Menschen mit M. Huntington oder die Menschen, die die Erkrankung bekommen werden, tragen eine “normale” und eine expandierte Wiederholungslänge. Nur eine sehr kleine Anzahl an Menschen trägt zwei verlängerte Kopien.
CAG-Blöcke mit 36 oder mehr Wiederholungen führen zur Erkrankung. Allerdings ist für Kopien zwischen 36 und 39 CAG eine reduzierte Penetranz (RP, siehe Abb. 4) bekannt, was bedeutet, dass nicht alle Menschen, die eine dieser CAG-Längen tragen, auch erkranken werden. Wenn sie erkranken, zeigen sie oft erst sehr spät die ersten Symptome.
Kopien zwischen 27 und 35 CAG sind nicht mit Krankheitssymptomen assoziiert; für Nachfahren besteht jedoch ein erhöhtes Risiko einer Expansion in den pathologischen Bereich. Diese Kopien mit intermediären Längen (IL, siehe Abb. 4) sind mit ein Grund dafür, dass sporadische Krankheitsfälle beobachtet werden, also bisher niemand sonst in der Familie erkrankt ist. Diese Fälle sind jedoch sehr selten.
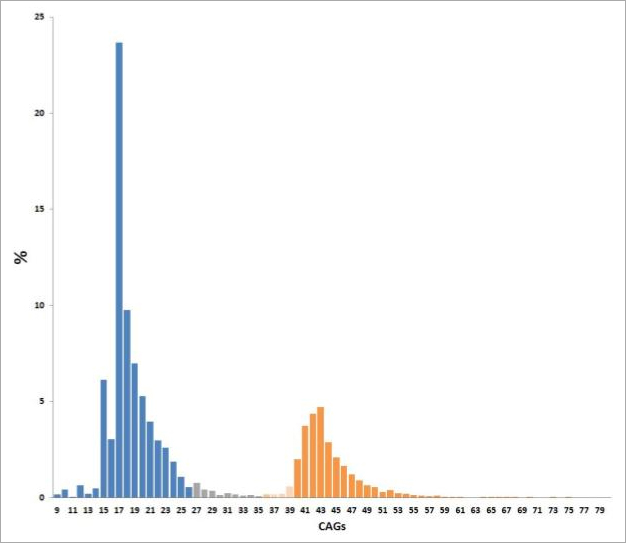
Abb. 3. Verteilung der CAG-Längen. “Normale” CAG-Längen (blau und grau) im Huntingtin-Gen umfassen bis zu 35 CAG-Wiederholungen. Am häufigsten finden sich 17 CAG-Wiederholungen. CAG-Blöcke mit 36 oder mehr Wiederholungen führen zur Erkrankung, wobei ab 40 Wiederholungen (orange) immer Symptome im Laufe des Lebens auftreten. In diesem Bereich finden sich am häufigsten Kopien mit 41-43 CAG-Wiederholungen.
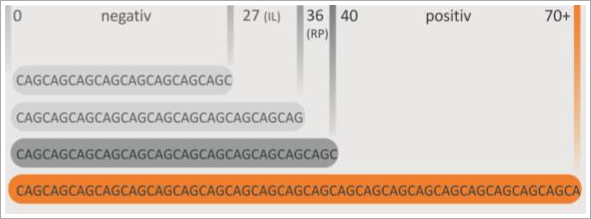
Abb. 4 Bei einer ermittelten CAG-Länge von unter 27-Wiederholungen spricht man von einem negativen Befund. CAG-Blöcke mit 40 oder mehr Wiederholungen (orange) führen ohne Ausnahme zur Erkrankung. Bei Kopien zwischen 36 und 39 CAG spricht man von reduzierter Penetranz (RP), nicht alle Menschen die eine dieser Längen tragen werden auch erkranken. Kopien mit sog. intermediären Längen (IL) zwischen 27 und 35 CAG sind nicht mit Krankheitssymptomen assoziiert; für Nachfahren besteht jedoch ein erhöhtes Risiko einer Expansion in den pathologischen Bereich.
Ein weiteres Merkmal der verlängerten CAG-Blöcke ist, dass ihre Länge mit dem Auftreten der ersten Symptome korreliert: je mehr CAG-Wiederholungen desto früher bricht die Erkrankung aus (Abb. 5). Die CAG-Länge erklärt allerdings nur ca. 50-70% der Varianz im Manifestationsalter, so dass von der individuellen CAG-Länge einer Person keine Rückschlüsse auf den genauen Zeitpunkt der Erkrankung gezogen werden können. Neben Umwelteinflüssen bestimmen auch weitere genetische Faktoren, ob die Erkrankung früher oder später ausbricht. Seit einigen Jahren beschäftigt sich eine Arbeitsgruppe des Huntington-Zentrums mit der Suche nach sogenannten M. Huntington-modifizierenden Genen.
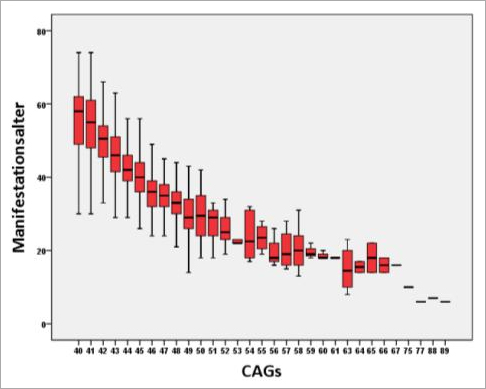
Abb. 5 Abhängigkeit des Huntington-Manifestationsaltes von der CAG-Länge.
Wann wird ein genetischer Test durchgeführt?
Bei der Testung auf die M. Huntington-Mutation wird die exakte CAG-Länge im Huntingtin-Gen bestimmt. Grundsätzlich lassen sich zwei Gründe für den Test unterscheiden: Neben der prädiktiven DNA-Diagnostik für Risikopersonen ohne Krankheitszeichen ist auch eine DNA-Untersuchung als Differentialdiagnostik möglich.
Im Fall der Differentialdiagnostik wird der Test durchgeführt, um eine eventuelle M. Huntington Diagnose in einem Patienten mit entsprechenden Symptomen zu bestätigen oder zu widerlegen.
Die genetische Untersuchung kann von jeder Ärztin und jedem Arzt nach Aufklärung gemäß §10 Gen-DG und schriftlicher Einwilligung vorgenommen werden. Da ein positives Ergebnis allerdings weitreichende Folgen für die ganze Familie haben kann, sollte in diesem Fall immer nach auf die Möglichkeit einer humangenetischen Beratung hingewiesen werden.
Für gesunde Angehörige von betroffenen Patienten ist eine prädiktive genetische Diagnostik möglich. Prädiktive genetische Untersuchungen dürfen nur von Fachärztinnen oder Fachärzten der Humangenetik oder andern Ärztinnen und Ärzten vorgenommen werden, die sich beim Erwerb einer Facharzt-, Schwerpunkt- oder Zusatzbezeichnung für genetische Untersuchungen im Rahmen ihres Fachgebiets qualifiziert haben (Gen-DG).
Ablauf einer genetischen Beratung zur prädiktiven Testung auf M. Huntington
Jede Person, die von sich glaubt, ein Risiko für M. Huntington zu haben, kann eine genetische Beratung in Anspruch nehmen. Ein internationales Gremium aus Fachleuten, betroffenen Patienten sowie deren Angehörigen (Selbsthilfegruppen) hat Richtlinien erarbeitet, nach denen die prädiktive Diagnostik in einem Zeitplan mit Mindestreflexionszeiten für jeden Beratungsabschnitt durchzuführen ist.
In einem Erstgespräch werden im Wesentlichen Informationen über die Erkrankung sowie deren Erbgang und die Erkrankungsrisiken informiert. Es wird eine ausführliche Familien- und Eigenanamnese erhoben. Bei dem ersten Termin werden auch insbesondere die Möglichkeiten, Aussagekraft und Konsequenzen der molekulargenetischen Diagnostik besprochen und die Bedeutung der Aussa-gen und der evtl. zu erhebenden Befunde für andere Familienmitglieder erläutert. Bei dem Erstgespräch erfolgt keine Blutentnahme.
Vor der Blutentnahme wird empfohlen, dass der/die Ratsuchende ein Gespräch mit einem Psychologen führt, der mit den Besonderheiten der Huntington Krankheit und der prädiktiven Diagnostik vertraut ist und auch nach der Diagnostik für weitere Gespräche zur Verfügung stehen würde. Im Erstgespräch können entsprechende Kontakte mit kooperierenden Psychologen vermittelt werden, die zeitnah Termine vergeben. Der/die psychotherapeutische Berater(in) sollte sich vergewissern, dass eine prädiktive Diagnostik auf der Grundlage umfassender Information vom Ratsuchenden gewollt wird, und dass vor allem eine ungünstige Diagnosemitteilung verarbeitet werden kann.
Zusätzlich wird der/die Ratsuchende gebeten sich eine Vertrauensperson zu wählen welche während der Vorbereitungsphase auf die Diagnostik sowie bei der Befundmitteilung und auch danach begleitet zur Seite steht. Bei der Vertrauensperson kann es sich um den/die Lebenspartner/-in, einen/eine Freund/-in oder irgendeine andere Person handeln, in welche der/die Ratsuchende das entsprechende Vertrauen hat. Die Vertrauensperson sollte aber möglichst nicht selbst Risikoperson sein.
Die Blutentnahme und anschließende molekulargenetische Untersuchung erfolgt dann, wenn die obengenannten Rahmenbedingungen gewährleistet sind und wenn die Risikoperson und der/die psychotherapeutische Berater(in) ihre Zustimmung gegeben haben.
Das Ergebnis wird an den Arzt/die Ärztin, der/die die genetische Beratung durchgeführt hat übermit-telt. Die Ergebnismitteilung durch das Labor in der Humangenetik des Zentrums erfolgt allerdings so, dass die beteiligten Berater/Beraterinnen selbst nicht über das Ergebnis informiert sind. Erst auf Wunsch des/der Ratsuchenden wird der Befund mit dem Ergebnis geöffnet und mitgeteilt.
Die Risikoperson kann jederzeit erklären, dass sie an der Fortsetzung der Untersuchung bzw. der Befundmitteilung nicht mehr interessiert ist. Auf ausdrücklichen Wunsch kann jederzeit die DNA-Probe vernichtet werden (Gen-DG).