Teilprojekt D: Prof. Dr. G. Jansen
Universität Duisburg-Essen
Quantenchemische Berechnungen molekularer Aggregate
Kleine und große molekulare Aggregate wie Cluster in der Gasphase und Molekülkristalle verdanken ihre Existenz dem Wechselspiel der elektrostatischen, Induktions-, Dispersions- und Austauschwechselwirkungen zwischen den Molekülen. Mit der DFT-SAPT Kombination aus intermolekularer Störungstheorie zur Beschreibung der Wechselwirkungen und Dichtefunktionaltheorie zur Berechnung der notwendigen molekularen Eigenschaften verfügen wir über eine effiziente Methode zur genauen Charakterisierung der Abstands- und Orientierungsabhängigkeit der diversen zwischenmolekularen Kräfte. Diese Methode wurde erfolgreich zum Studium der Wechselwirkungen zwischen Molekülen mit CH-Gruppen, p-Systemen und einsamen Elektronenpaaren (Acetylen, Benzol, Heteroaromaten) und ihrer Konsequenzen auf Bildung und Strukturen molekularer Aggregate eingesetzt.
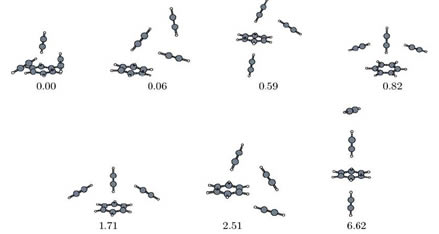
Verschiedene Strukturen des Aggregats von einem Benzol- und drei Acetylenmolekülen, erhalten mit DFT-SAPT-basierten Modellpotentialen. Relative Energien in kJ/mol.
Die Auswirkungen der Substitution von CH- durch CF-Gruppen bildet einen neuen Fokus unserer Untersuchungen, wie auch in entsprechenden experimentellen Arbeiten anderer Projekte von FOR618. DFT-SAPT und weitere quantenchemische Methoden werden dazu benutzt, Wechselwirkungsenergien zu berechnen, die Wechselwirkungen über die einzelnen Energiebeiträge zu charakterisieren und analytische Darstellungen der Potentialenergieflächen der wechselwirkenden Moleküle zu gewinnen. Diese bilden dann die Basis von Molekulardynamik-Simulationen von Clustern und periodischen Systemen sowie der Berechnung von Vibrations-Rotations-Tunnelspektren, was den direkten Vergleich mit dem Experiment ermöglicht.