aRNA-longSAGE
Heidenblut AM, Luttges J, Buchholz M, Heinitz C, Emmersen J, Nielsen KL, Schreiter P, Souquet M, Nowacki S, Herbrand U, Kloppel G, Schmiegel W, Gress T, Hahn SA. ![]() aRNA-longSAGE: a new approach to generate SAGE libraries from microdissected cells. Nucleic Acids Res. 2004 Sep 15;32(16):e131. PMID: 15371555
aRNA-longSAGE: a new approach to generate SAGE libraries from microdissected cells. Nucleic Acids Res. 2004 Sep 15;32(16):e131. PMID: 15371555
The detailed aRNA-longSAGE protocol ![]() and representative example figures can be downloaded here (no of downloads:
).
and representative example figures can be downloaded here (no of downloads:
).
Information on aRNA-longSAGE in German language ![]() can be obtained from the dissertation of A.M. Heidenblut.
can be obtained from the dissertation of A.M. Heidenblut. ![]()
INTRODUCTION
With the availability of histopathologically defined tumor progression models it has become of key interest to identify important cell biological changes that are responsible for the development of the various tumor progression stages. Some insights came from the identification of activated oncogenes or inactivated tumor suppressor genes. To understand how these activated or inactivated tumor genes alter the complex cellular signaling and thus drive tumor progression, gene expression analyses of normal cells and their corresponding carcinoma precursor and carcinoma cells are crucial. Currently, gene expression analyses at the level of single candidate genes or broader expression analysis approaches, such as serial analysis of gene expression (SAGE) and microarray technology, are being used to reach this goal. It is expected that this knowledge will ultimately help not only to define new therapeutic target genes and prognostic gene expression patterns but also to identify new (early) diagnostic markers.
To be able to analyze the expression profile of distinct histological cell types within a complex primary tissue, a method to isolate the cells of interest is needed. Microdissection using laser capture or manual techniques has been successfully used to produce such highly enriched cell preparations. Because the number of cells available through microdissection is limited in most instances, the amount of RNA that can be obtained from these samples is not sufficient for standard gene expression profiling protocols. In order to generate gene expression profiles from microdissected cells it is necessary to amplify the amount of starting material, either by T7-based RNA amplification or by polymerase chain reaction (PCR) amplification of the cDNA. The linear amplification of RNA by in vitro transcription, as introduced by Eberwine et al. (1), has been shown to result in less amplification bias than the PCR amplification of cDNA (2) and has been successfully applied for the gene expression profiling of microdissected cells using microarray technology (3-5).
An inherent limitation of microarrays is their ability to identify only predefined transcripts present on the array. SAGE, in turn, is a powerful alternative for performing expression analyses without prior knowledge of the genes to be identified (6). This technique creates gene expression profiles by generating libraries of short cDNA sequence tags, each tag representing an mRNA transcript. Via concatenation, cloning and high throughput sequencing of the tags the gene expression profile is generated (6). SAGE libraries have been widely used to study the genetic changes underlying the transformation from normal to cancer cells (for review see (7)). A systematic analysis of publicly available SAGE tags has recently shown that a significant proportion of tags likely represent unknown genes or new isoforms of known genes, indicating that SAGE is truly complementary to current microarray technology (8). In addition, the recent introduction of longSAGE, a SAGE variant that produces 21 bp tags instead of the 14 bp tags obtained from conventional SAGE libraries, further increased the reliability of SAGE-tag-sequence to gene annotation. In addition, the 21 bp longSAGE tags can be used directly as primers for the isolation of novel transcripts using PCR technology (9).
Only few modifications of the current SAGE technology have so far been published that enable SAGE to be applied to less than 5 x 104 cells, a prerequisite for using SAGE for microdissected tissues. Two methods, PCR-SAGE (10) and SAGE-lite (11), rely on PCR amplification of the cDNA at the beginning of the SAGE procedure. A third method described by Schober et al. (12) requires an additional ditag PCR-amplification step. In all cases PCR amplification is likely to introduce a bias in the resulting expression profile (2,13).
The recently published small amplified RNA-SAGE approach (14) uses a modified protocol for T7-based RNA amplification that, in contrast to the Eberwine protocol, yields amplified sense RNA. The amplified RNA is then processed according to the standard SAGE protocol.
Here we present a protocol starting with microdissection, followed by a modification of the SAGE protocol which is the first to allow the direct use of amplified antisense RNA (aRNA) generated by means of the well established and validated Eberwine protocol for SAGE library generation.
MICRODISSECTION AND aRNA-PRODUCTION
To generate dedicated expression profiles of microscopic lesions it is mandatory to apply microsdissection to enrich for the target cell population. In our hands the manual microdissection procedure proved to be easier and speedier than using a laser capture microdissection system (Arcturus PixCell II) available at our institute. Figure 1 ![]() shows a representative example of the quality and length for RNA and amplified antisense RNA derived from microdissected cells generated with an RNA 6000 Pico LabChip®on a Bioanalyzer platform.
shows a representative example of the quality and length for RNA and amplified antisense RNA derived from microdissected cells generated with an RNA 6000 Pico LabChip®on a Bioanalyzer platform.
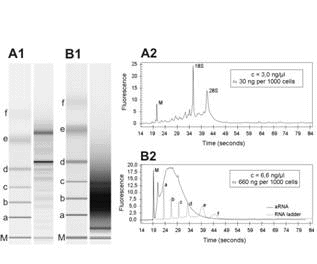
Figure 1: Analysis of RNA quality and length for RNA derived from microdissected cells. Representative RNA gel images generated with an RNA 6000 Pico LabChip®on a Bioanalyzer platform are shown for total RNA (A1) and aRNA (B1). Corresponding electropherograms are shown for total RNA (A2) and aRNA (B2). M, marker for sample synchronization; 18S, 18S rRNA; 28S, 28S rRNA; a-f, RNA ladder (0.2, 0.5, 1.0, 2.0, 4.0, 6.0 kb).
SAGE it was previously not possible to use the aRNA produced by the available standard protocols directly as starting material. aRNA-longSAGE overcomes this limitation. To be able to use linearly amplified aRNA as starting material for the generation of SAGE libraries, we modified the cDNA synthesis steps in the SAGE protocol as follows (see also Fig.2 ![]() ): First of all the aRNA was reverse transcribed with a random primer that included the recognition site of the SAGE anchoring enzyme NlaIII (SAGE-random primer 5´-NNN NNN CAT G-3´). The recognition site for NlaIII was introduced, because we wanted to specifically enrich for target sequences that are needed for subsequent steps in the SAGE protocol. The RNA was then removed from the resulting DNA-RNA hybrid by digestion with RNase H. At this point the cDNA first strand corresponds to the mRNA before amplification and has a 3´polyA tail which can be annealed to magnetic oligo(dT) beads. After coupling of the first strand synthesis product to the magnetic beads, the (dT)25 oligonucleotide linked to the beads served as primer for the second strand cDNA synthesis step. The resulting cDNA can be used directly in either the conventional MicroSAGE protocol or the MicroSAGE protocol modified for longSAGE (9).
): First of all the aRNA was reverse transcribed with a random primer that included the recognition site of the SAGE anchoring enzyme NlaIII (SAGE-random primer 5´-NNN NNN CAT G-3´). The recognition site for NlaIII was introduced, because we wanted to specifically enrich for target sequences that are needed for subsequent steps in the SAGE protocol. The RNA was then removed from the resulting DNA-RNA hybrid by digestion with RNase H. At this point the cDNA first strand corresponds to the mRNA before amplification and has a 3´polyA tail which can be annealed to magnetic oligo(dT) beads. After coupling of the first strand synthesis product to the magnetic beads, the (dT)25 oligonucleotide linked to the beads served as primer for the second strand cDNA synthesis step. The resulting cDNA can be used directly in either the conventional MicroSAGE protocol or the MicroSAGE protocol modified for longSAGE (9).
{kind=link}
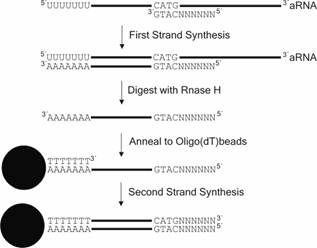
Figure 2: Scheme of the modified cDNA synthesis protocol within the aRNA-long-SAGE procedure.
THE SAGE PROCEDURE
The standard longSAGE procedure is depicted in Figure 3 ![]() . The first step of any SAGE protocol requires the restriction of the cDNA with the “anchoring” enzyme. Usually frequent cutters like NlaIII are used because they are likely to cut in average every 400 bp within most cDNA molecules. This is important to ensure a high chance to have a successful cut ideally within the 3-prime end of the majority of transcripts represented in the cDNA pool under study. Following this enzymatic step the cDNA is divided into two aliquots and different linkers are ligated to the ends of each pool of cDNAs. Each linker contains a different PCR-primer binding sequence and a recognition site for a class IIS restriction endonuclease (i.e. MmeI for longSAGE), also called “tagging” enzyme. The important feature of the “tagging” enzyme is its ability to cut several bps away from its recognition site (in the case of MmeI in average 20 bp). In the next step tags containing a short sequence of the transcript are released by cleaving each cDNA pools with the “tagging” enzyme. Now the two pools of tags are ligated to each other forming “ditags”. Next, the only PCR amplification step within the SAGE protocol is performed using the primer set complementary to the primers sequences from the linkers. This PCR step ensures enough product for the subsequent concatenization and ligation step prior to en masse sequencing of the inserts.
. The first step of any SAGE protocol requires the restriction of the cDNA with the “anchoring” enzyme. Usually frequent cutters like NlaIII are used because they are likely to cut in average every 400 bp within most cDNA molecules. This is important to ensure a high chance to have a successful cut ideally within the 3-prime end of the majority of transcripts represented in the cDNA pool under study. Following this enzymatic step the cDNA is divided into two aliquots and different linkers are ligated to the ends of each pool of cDNAs. Each linker contains a different PCR-primer binding sequence and a recognition site for a class IIS restriction endonuclease (i.e. MmeI for longSAGE), also called “tagging” enzyme. The important feature of the “tagging” enzyme is its ability to cut several bps away from its recognition site (in the case of MmeI in average 20 bp). In the next step tags containing a short sequence of the transcript are released by cleaving each cDNA pools with the “tagging” enzyme. Now the two pools of tags are ligated to each other forming “ditags”. Next, the only PCR amplification step within the SAGE protocol is performed using the primer set complementary to the primers sequences from the linkers. This PCR step ensures enough product for the subsequent concatenization and ligation step prior to en masse sequencing of the inserts.
{kind=link}
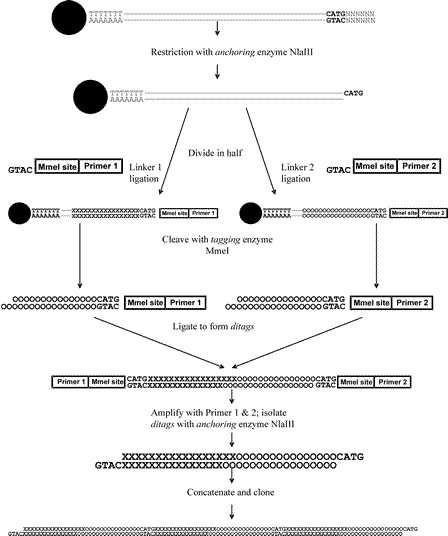
Figure 3: Scheme of the standard long-SAGE procedure.
Following sequencing, longSAGE tags can be extracted from the sequence files with the SAGE-PHRED 2003 software (can be obtained from je@bio.auc.dk ) or the SAGE 2000 software (http://www.sagenet.org/ ![]() ). To keep the sequencing error rate as low as possible it is advisable to set the minimum quality of each base within a tag sequence to PHRED20 and to set the maximum ditag length to 36 (flanking CATGs not included). Generally, some 50.000 tags are collected for each library, because it has previously been shown that above this number of tags the chance to identify new tag sequences is dropping dramatically thus the sequencing effort is rising disproportional (6). For the tag to gene annotation a number of tools/databases are available such as the “SAGEmap_tag_ug-rel” database (ftp://ftp.ncbi.nlm.nih.gov/pub/sage
). To keep the sequencing error rate as low as possible it is advisable to set the minimum quality of each base within a tag sequence to PHRED20 and to set the maximum ditag length to 36 (flanking CATGs not included). Generally, some 50.000 tags are collected for each library, because it has previously been shown that above this number of tags the chance to identify new tag sequences is dropping dramatically thus the sequencing effort is rising disproportional (6). For the tag to gene annotation a number of tools/databases are available such as the “SAGEmap_tag_ug-rel” database (ftp://ftp.ncbi.nlm.nih.gov/pub/sage ![]() ), as of August 2004 it contained 795,885 entries) or the web portal “SAGE Genie” (http://cgap.nci.nih.gov/SAGE
), as of August 2004 it contained 795,885 entries) or the web portal “SAGE Genie” (http://cgap.nci.nih.gov/SAGE ![]() ). Of note, to further correct for potential sequencing errors, only tag sequences identified twice are usually regarded as reliable and therefore processed in subsequent analyses. For a comparison between two libraries to generate a differential expression profile, normalization of the SAGE library needs to be performed to correct for differences in the overall tag sum per library. Subsequently, differentially expressed genes can be identified by statistical means, i.e. with the help of the program SAGEstat which performs a Z-test (15).
). Of note, to further correct for potential sequencing errors, only tag sequences identified twice are usually regarded as reliable and therefore processed in subsequent analyses. For a comparison between two libraries to generate a differential expression profile, normalization of the SAGE library needs to be performed to correct for differences in the overall tag sum per library. Subsequently, differentially expressed genes can be identified by statistical means, i.e. with the help of the program SAGEstat which performs a Z-test (15).
REFERENCES
- Eberwine, J., Yeh, H., Miyashiro, K., Cao, Y., Nair, S., Finnell, R., Zettel, M. and Coleman, P. (1992) Proc Natl Acad Sci U S A, 89, 3010-3014.
- Puskas, L.G., Zvara, A., Hackler, L., Jr. and Van Hummelen, P. (2002) Biotechniques, 32, 1330-1334, 1336, 1338, 1340.
- Kitahara, O., Furukawa, Y., Tanaka, T., Kihara, C., Ono, K., Yanagawa, R., Nita, M.E., Takagi, T., Nakamura, Y. and Tsunoda, T. (2001) Cancer Res, 61, 3544-3549.
- Grutzmann, R., Foerder, M., Alldinger, I., Staub, E., Brummendorf, T., Ropcke, S., Li, X., Kristiansen, G., Jesnowski, R., Sipos, B. et al. (2003) Virchows Arch, 443, 508-517.
- Ohyama, H., Zhang, X., Kohno, Y., Alevizos, I., Posner, M., Wong, D.T. and Todd, R. (2000) Biotechniques, 29, 530-536.
- Velculescu, V.E., Zhang, L., Vogelstein, B. and Kinzler, K.W. (1995) Science, 270, 484-487.
- Polyak, K. and Riggins, G.J. (2001) J Clin Oncol, 19, 2948-2958.
- Chen, J., Sun, M., Lee, S., Zhou, G., Rowley, J.D. and Wang, S.M. (2002) Proc Natl Acad Sci U S A, 99, 12257-12262.
- Saha, S., Sparks, A.B., Rago, C., Akmaev, V., Wang, C.J., Vogelstein, B., Kinzler, K.W. and Velculescu, V.E. (2002) Nat Biotechnol, 20, 508-512.
- Neilson, L., Andalibi, A., Kang, D., Coutifaris, C., Strauss, J.F., 3rd, Stanton, J.A. and Green, D.P. (2000) Genomics, 63, 13-24.
- Peters, D.G., Kassam, A.B., Yonas, H., O'Hare, E.H., Ferrell, R.E. and Brufsky, A.M. (1999) Nucleic Acids Res, 27, e39.
- Schober, M.S., Min, Y.N. and Chen, Y.Q. (2001) Biotechniques, 31, 1240-1242.
- Ye, S.Q., Zhang, L.Q., Zheng, F., Virgil, D. and Kwiterovich, P.O. (2000) Anal Biochem, 287, 144-152.
- Vilain, C., Libert, F., Venet, D., Costagliola, S. and Vassart, G. (2003) Nucleic Acids Res, 31, E24.
- Ruijter, J.M., Van Kampen, A.H. and Baas, F. (2002) Physiol Genomics, 11, 37-44.