![]()
![]()
![]()
![]()
The projects not actively pursued anymore. We provide the last mature version if possible, but adoption to current programming language / library versions or user requests and question support is not guaranteed.
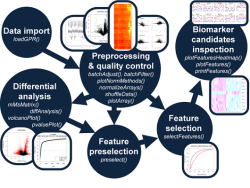
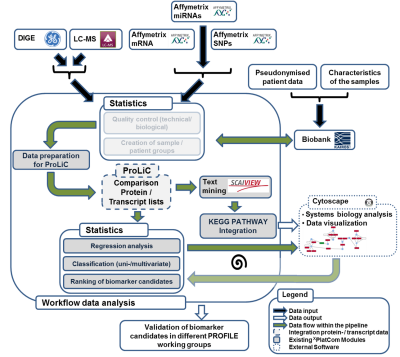
Prevention and personalized medicine are key issues of contemporary medical research. Multi-OMICS approaches aim at measuring the dynamics of the most important biomolecules (i.e. genes, mRNAs, proteins and metabolites) in order to gain better understanding of the complex regulation of a cell. In the medical context, such efforts are promising for the discovery of novel biomarkers and the development of new drug targets. However, processing and interpretation of multi-OMICS data is usually challenging and requires a structured workflow (Fig. 1).
Within the multi-OMICS pro-ject named PROFILE data sets obtained from several high - throughput technology platforms (Transcriptomics, Proteomics, Epigenetics, cirulating tumor cells) are analysed. For this data processing a standardized workflow has been developed (Fig.1), which comprises several steps of data conversion, quality control, data comparison, text mining and statistical analyses. Additionally, a software named CrossPlatformCommander (XPlatCom) has been programmed, which facilitates several steps of the proposed workflow in a semi-automatic manner. XPlatCom is currently in development (Beta status).
A more detailed description of the software is given by the poster available for download below and by the literature (PMID:23501674).
Figure 1: Sketch of the data processing workflow within the PROFILE project. Note, that transparent parts of the sketch indicate data processing steps, which will be implemented within XPlatCom in the near future.
In the recent years, high throughput proteomics has been gained remarkable progress resulting in huge data sets of protein identifications obtained from different cells or tissues. This often heterogeneous and distributed information needs to be integrated and compared in order to achieve a more comprehensive understanding of the cell function.
However, a direct comparison of available data, i.e. the protein lists along with related information (e.g. accession numbers, amino acid sequences, the assigned peptide identifications etc.), is challenging. For example, protein accessions may be obtained from various databases with different nomenclature. Furthermore, even within the same database, accession numbers may change with different database versions. Therefore, matching of protein identifications with respect to accession numbers alone is usually unsatisfying.
Furthermore, the assignment of experimentally obtained spectrum information to peptide sequences and the subsequent protein inference, usually contains uncertainty (e.g. due to an incomplete sequence coverage or an ambiguous peptide pattern).
The Protein List Comparator (ProLiC) software addresses these issues. To this end, ProLiC includes different possibilities to solve the problem of protein list comparison:
In addition to a simple accession based comparison, which is still supported by the software, ProLiC permits a more reliable comparison of protein lists from different sources by considering measured sequence and/or peptide information. The above mentioned uncertainty is addressed by the development of a protein grouping algorithm along with an adequate definition of a protein similarity criteria. The protein grouping algorithm of ProLiC is highly customizable via parameter settings given in the configuration file of the software.
ProLiC is a tool that can be used from the command line, which enables incorporation of ProLiC comparisons in ordinary shell scripts. The software is open source, free of charge and can be downloaded using the download link below. The download includes a compiled version of the program, the sources, a HTML documentation created with Javadoc as well as a detailed manual. In addition a workflow for the KNIME workflow engine is provided, which allows conversion of Proteome Discoverer result files into the ProLiC format.
The Proteomics Conversion Tool was prototyped within the EU funded "Coordination Action" ProDaC (grant number LSHG-CT-2006-036814). New features were added within the EU funded "Collaborative Project" ProteomeXchange (grant number 260558).
ProCon is a Java application for conversion of data from Proteomics files or a LIMS (Laboratory Information Management System) database into standard formats.
ProCon allows the conversion of:
For more details about ProCon please download our poster or the ProCon paper (coming soon).
ProCon 0.9.804 needs Java 13 or higher.
Download the proper zip package, unzip it (e.g.to C:\ProCon_dist-0.9.xyz) and follow the user installation and configuration instructions in .\documentation\ProCon_user_manual.pdf.
The mpc Statistical DIGE Analyzer is a Java-application for the statistical analysis of data from 2-Dimensional Difference Gel Electrophoresis (DIGE). It was developed by Dr. Klaus Jung at the Medizinisches Proteom-Center (Prof. Dr. Helmut E. Meyer) of the Ruhr-Universität Bochum. The software comprises the following features:
The mpc Statistical DIGE Analyzer is a Java-application for the statistical analysis of data from 2-Dimensional Difference Gel Electrophoresis (DIGE). It was developed by Dr. Klaus Jung at the Medizinisches Proteom-Center (Prof. Dr. Helmut E. Meyer) of the Ruhr-Universität Bochum. The software comprises the following features:
In order to use this application download the .rar-file below and unpack it to the desired directory of your computer. The .rar-file can be unpacked by using WinRAR. Start the application by opening the SDA.jar-file in the main folder.
In regular proteomics approaches proteases are used to digest the proteome into a set of peptides. Unfortunately, determining proteins on the basis of peptides implies some uncertainty since a peptide may be part of different proteins. Therefore, a targeted detection of unique peptides of particular proteins is a promising task for an unambiguous identification of a specific protein.
The Unique Peptide Finder (UPF) software offers the possibility for a highly efficient and simple detection of such unique peptides. In a first step a SQL-based database of theoretically digested peptides from a given FASTA file formatted protein database is generated by choosing a protease. In a second step, in silico generated peptides from a pre-defined protein sequence are compared to this peptide database in order to identify unique peptides. Amongst others, possible applications are identification of proteins when only sparse peptide information is available or advanced proteomics techniques that require information about the uniqueness of peptides such as Multiple Reaction Monitoring (MRM).
A detailed exemplification of the software and the application of UPF using the example of several isoforms of the human Cytochrome P450 (CYP) family is given by Kohl M., Redlich G., Eisenacher M., Schnabel A., Meyer H.E., Marcus K. and Stephan C. (2008) Automated Calculation of Unique Peptide Sequences for Unambiguous Identification of Highly Homologous Proteins by Mass Spectrometry. Journal of Proteomics and Bioinformatics Vol. 1, 6-10 (http://www.omicsonline.com/Archive/HTMLApril2008/JPB1.6.html).
DecoyDatabaseBuilder is a module of the PeakQuant suite, it enables the generation of decoy sequences for amino acid databases.
It is written in Java (version 1.5.x or higher is required for Microsoft Windows, Java 1.6.x for Linux and Apple).
At the moment the DecoyDatabaseBuilder software is publicly available and free for non-commercial use.
Hint: Users encountered fatal exceptions when using 32-bit versions of Java; you should try 64-bit also (don't forget to change the browser's helper application for JNLP files to this 64-bit version)!
If you use DecoyDatabaseBuilder to publish results please cite the following reference:
"Reidegeld KA, Eisenacher M, Kohl M, Chamrad D, Körting G, Blüggel M, Meyer HE, Stephan C (2008): An easy-to-use Decoy Database Builder software tool, implementing different decoy strategies for false discovery rate calculation in automated MS/MS protein identifications. Proteomics 2008 Mar;8(6):1129-37" [PubMed ID: 18338823]
At the moment the DecoyDatabaseBuilder software is publicly available and free for non-commercial use.
To use DecoyDatabaseBuilder as local installed version, download the following file, unzip to an arbitrary folder like "C:\DecoyDatabaseBuilder \" and double-click DecoyDatabaseBuilder .jar:
PeakQuant (formerly known as Peakardt) serves as an integrated platform for several Proteomics tools and provides an easily operated graphical user interface.
One important module is FindPairs - a algorithm aiming on automated quantitative analysis of isotope-labelled mass spectra with high accuracy and reliability (e.g. SILAC and 14N/15N metabolic labelling).
Other important modules are DecoyDatabaseBuilder and SetOperations, each also available as separate executables.
PeakQuant is written in Java (version 1.5.x or higher is required for Microsoft Windows, Java 1.6.x for Linux and Apple computers, 64bit versions enabling more than 3.2 GB memory are recommended!).
At the moment the PeakQuant software suite is publicly available and free for non-commercial use.
To use PeakQuant as local installed version, download the following file, unzip to an arbitrary folder like "C:\PeakQuant\" and double-click PeakQuant.jar or PeakQuant.bat:
If you use PeakQuant to publish results, please cite the following reference:
"Eisenacher M, Kohl M, Wiese S, Hebeler R, Meyer HE, Warscheid B, Stephan C (2012) Find Pairs: The Module for Protein Quantification of the PeakQuant Software Suite. OMICS: A Journal of Integrative Biology 2012 Sep;16(9):457-67." [PubMed ID: 22909347]
14N / 15N test data set (mzXML converted excerpt of spectra set used in Hebeler et al. (2008) - spot 58, experiment 1, glutathione S-transferase) and suitable configuration files:
SetOperations is a module of the PeakQuant suite, it allows the set operations "union", "intersection", and "difference" on up to 23 accession lists / sets.
It is written in Java (version 1.5.x or higher is required for Microsoft Windows, Java 1.6.x for Linux and Apple computers).
At the moment the SetOperations software is publicly available and free for non-commercial use.
To start DecoyDatabaseBuilder using the Java webstart mechanism, press the following link:
At the moment the SetOperations software is publicly available and free for non-commercial use.
To use SetOperations as local installed version, download the following file, unzip to an arbitrary folder like "C:\SetOperations\" and double-click SetOperations.jar:
The mzIdentML-Validator is a software tool of the HUPI-PSI for the semantic validation of mzIdentML files.
PAA is an R/Bioconductor software tool for protein microarray data analysis aimed at biomarker discovery
A highly useful and flexible tool for calibration of targeted MS‐based measurements. CalibraCurve enables an automated batch-mode determination of dynamic linear ranges and quantification limits for both targeted proteomics and similar assays. The software uses a variety of measures to assess the accuracy of the calibration and provides intuitive visualizations. CalibraCurve was originally developed by Dr Michael Kohl and secondarily supervised by Dr. Markus Stepath.

Copyright © MPC 2023
Last update: May 30, 2023
{kind=link}
{kind=link}
{kind=link}
{kind=link}