intracellulärer membrantransport und Molekulare Mechanismen der Zellpolarisation
Unsere Arbeitsgruppe beschäftigt sich mit den molekularen Mechanismen der Zellpolarisation. Die bekanntesten Beispiele zellulärer Polarisation finden sich bei Epithel- sowie bei Nervenzellen. Epithelzellen verfügen über eine apikale, d.h. in der Regel dem Lumen zugewandte Domäne sowie eine basolaterale Domäne. Neurone hingegen zeigen hinsichtlich ihrer Neuriten eine Differenzierung in Axone und Dendriten. Störungen der Zellpolarisation sind häufig bei der Entartung von Zellen (Tumorbildung) zu beobachten und tragen zu deren Metastasierungs-Potential bei. Die genauen molekularen Mechanismen, die zur Ausbildung zellulärer Polarisation führen, sind weitgehend unklar. In unserer Arbeitsgruppe untersuchen wir in diesem Zusammenhang insbesondere die Funktion der Proteintyrosinphosphatase PTP-BL (ein Protein das in Epithelzellen apikal vorliegt) und den durch dieses Protein vermittelten supramoleularen Proteinkomplex.
| Vereinfachter schematischer Aufbau einer Epithelzelle mit den Proteinen PTP-BL an der apikalen und FRMPD2 an der basolateralen Membran. Dargestellt sind bisher identifizierte Interaktionspartner der PTP-BL und FRMPD2, welche an der Polarisation von Epithelzellen beteiligt sind. | 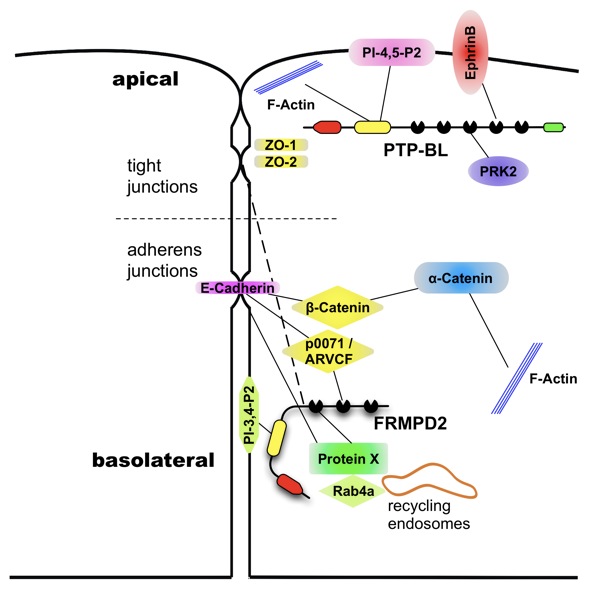 |
Molekulare Mechanismen des Lowe Syndroms- Die Funktion der Inositol-5-Phosphatase OCRL1
Das Lowe-Syndrom ist eine X-chromosomale Erbkrankheit, die durch Defekte des Nervensystems, der Augen sowie der Niere charakterisiert ist. An Lowe-Syndrom erkrankte Kinder werden bereits mit getrübten Linsen geboren, die in der Regel operativ entfernt werden müssen, zudem ist eine Hypotonie (geringe Muskelspannung) zu beobachten. Relativ früh nach der Geburt tritt ein dem Fanconi-Syndrom ähnlicher Defekt in den Nieren auf, der sich mit fortschreitendem Lebensalter verstärkt. Die Entstehung des Lowe-Syndroms kann auf Mutationen im Gen für das Protein OCRL zurückgeführt werden. Kürzlich konnte zudem gezeigt werden, dass Mutationen im OCRL Gen auch für das Auftreten des Dent II-Syndroms (bei einem Teil der Patienten) verantwortlich ist. Das Dent II-Syndrom ähnelt dem Lowe Syndrom, beschränkt sich aber im Wesentlichen auf einen Nierendefekt ohne einen weiteren okularen oder neuronalen Phänotyp. OCRL ist eine Inositol-5-Phosphatase, die selektiv die Position fünf am Inositolring von Phosphatidylinositolen dephosphorylieren kann. Inositolphosphate und Phosphatidylinositole spielen eine entscheidende Rolle in der Regulation des intrazellulären Vesikeltransports und in der Signaltransduktion. Eine aus der Funktion der OCRL abgeleitete molekulare Erklärung des im Lowe-Syndrom (bzw. Dent II-Syndrom) beobachteten Phänotyps existiert zurzeit nicht.
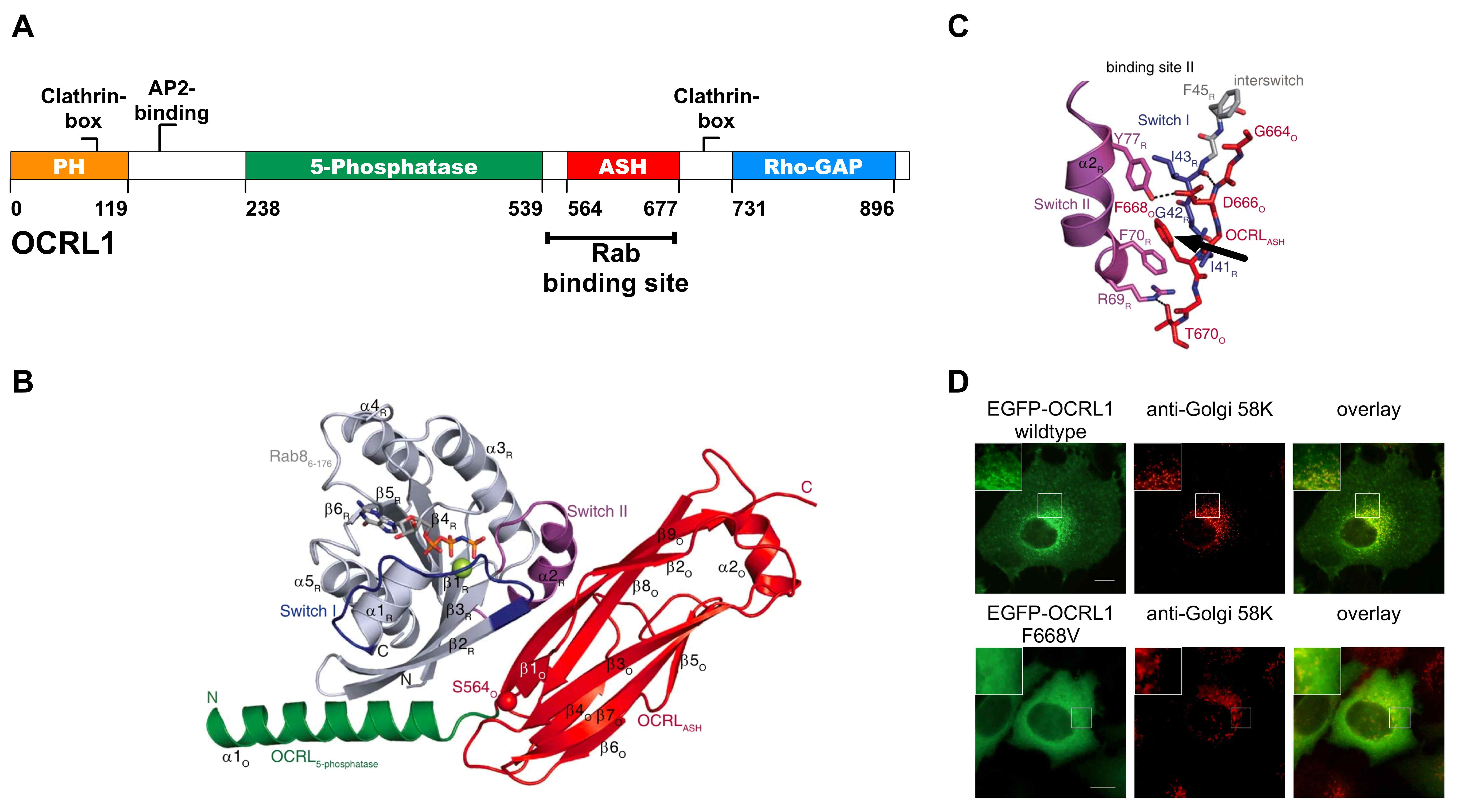 |
| A: Modularer Aufbau der OCRL1 bestehend aus einer N-terminalen Pleckstrin Homologie-Domäne (PH), einer zentralen Phosphatidylinositol-5-Phosphat-Phosphatase-Domäne gefolgt von einer ASPM/SPD2/Hydin-Domäne (ASH) und der C-terminalen Rho-GAP-ähnlichen-Domäne. OCRL1 verfügt über zwei Clathrin-Interaktionsstellen, ein AP2-Bindemotiv, sowie eine Rab-Bindestelle. B: Kristallstruktur der OCRL1 (Aminosäuren 540-678, grün und rot) im Komplex mit Rab8a (grau und lila) zeigt zwei Bindestellen; zum einen zwischen dem C-terminalen Abschnitt der Phosphatase-Domäne der OCRL1 und der Switch I bzw. der Interregion von Rab8a und zum anderen zwischen der ASH-Domäne der OCRL1 mit der Switch I und Switch II-Region von Rab8a. C: Detailansicht der zweiten Bindestelle verdeutlicht die Position des F668 (Pfeil) der OCRL1, welches in eine hydrophobe Tasche hineinragt, die aus den Aminosäuren I41, G42 und F70 des Rab8a gebildet wird.D: Vergleich der subzellulären Lokalisation von EGFP-OCRL1 (Wildtyp) mit EGFP-OCRL1, welche eine im Lowe-Syndrom auftretenden Mutation der Aminosäure F668 zu Valin aufweist, demonstriert eine veränderte Verteilung des Proteins innerhalb der Zelle. Während EGFP-OCRL1 Wildtyp in HeLa-Zellen vorwiegend am Golgi-Apparat und an endosomalen Strukturen lokalisiert ist, führt die Punktmutation F668V zu einer hauptsächlich cytosolischen Lokalisation; Maßstab entspricht 10 µm. |